Zuo-Guang. Ye Advanced Dielectric Piezoelectric and Ferroelectric Materials: Synthesis, Characterisation and Applications
Подождите немного. Документ загружается.

Handbook of dielectric, piezoelectric and ferroelectric materials1010
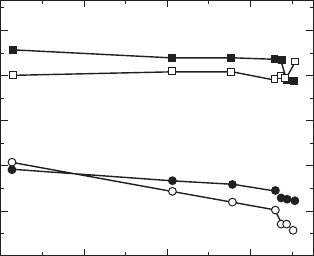
temperature calculated from the structural data determined by neutron
diffraction study. The V
Ti2
is 4.2 in the paraelectric state at 700°C, and
therefore Ti2 is overbonded in the octahedra. This indicates that the Ti2–O
bonds are too short and that the Ti2O
6
octahedra are under compressive
stress. The ferroelectric phase transition results in the V
Ti2
of approximately
4.0. This value is in good agreement with the nominal charge of Ti
4+
, indicating
that Ti2 is positioned in the crystallographically moderate environment in
the Ti2O
6
octahedra in the ferroelectric state. In contrast, the ferroelectric
transition leads to an increase in V
Ti1
from 4.0 (700°C) to 4.3 (25°C). Although
the V
Ti1
of 4.3 (25°C) is slightly overbonded, the difference from the nominal
charge (+4) is smaller than that of Bi.
The ferroelectric phase transition is found to result in a marked change in
V
Bi1
and V
Bi2
. In the paraelectric state, Bi1 and Bi2 are characterized by
strong underbonding. In particular, the underbonding of Bi1 is remarkable,
since V
Bi1
is only 2.3 at 700°C. This is much lower than the nominal charge
of Bi
3+
. This bond valence analysis exhibits that Bi1 in the paraelectric state
requires tighter bonding with oxide ions at the perovskite A site to satisfy
bonding requirement. The ferroelectric transition leads to an increase in V
Bi1
and V
Bi2
. The values of V
Bi1
and V
Bi2
are 3.0 at 25°C, which agrees well with
the nominal charge of Bi
3+
. While Bi1 cannot get a tight bonding with oxide
ions in the paraelectric state due to the restricted movement only along the
c-axis in the I4/mmm structure, the descending symmetry due to the ferroelectric
transition provides a three-dimensional degree of freedom for the displacements
of Bi1 and Bi2 to fulfill bonding requirement. The satisfactory bonding of
Bi1 and Bi2 stabilizes the ferroelectric distortion in the crystal. It is strongly
suggested that the loose bonding of Bi in the paraelectric state, especially the
Temperature /°C
6004002000
4.5
4.0
3.5
3.0
2.5
2.0
Bond valence sum,
V
Bi1
Bi2
Ti2
Ti1
33.3
Temperature dependence of bond valence sum (
V
) estimated
from the structural data.
V
represents static charge of ions in
crystals.
WPNL2204
Crystal structure and defect control 1011
underbonded Bi1 at the A site acts as the structural trigger for the ferroelectric
transition in the BiT system.
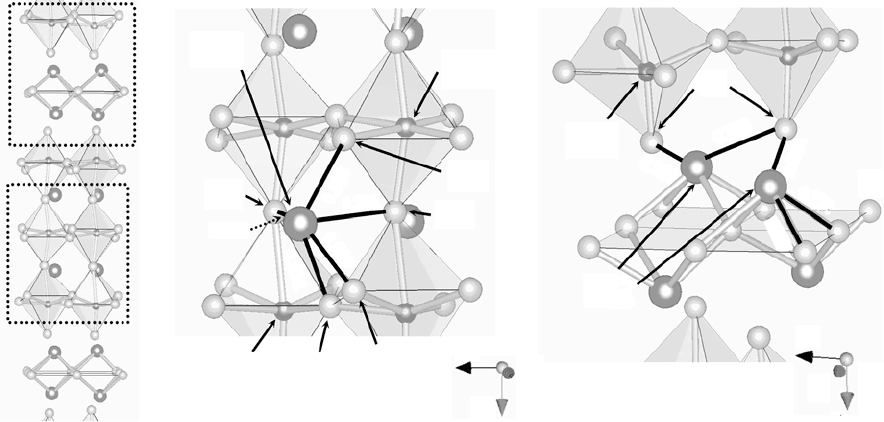
Figure 33.4 depicts the detailed bond length around Bi1 and Bi2 in the
ferroelectric state at 25°C. In the paraelectric state, Bi is located at the high-
symmetry 4e site. The ferroelectric transition leads to several short bonds as
shown in Fig. 33.4(b), and the satisfactory bonding of Bi1 is established. The
shortest bond is seen in bond length (l)
between Bi1 and O3 of 0.2284 along
the a-axis, which is 0.0458 nm (17%) smaller than that in the paraelectric
state. The strong bonding of Bi1–O3 plays an essential role in the ferroelectric
polarization as well as in the appropriate bond valence of the Bi1 at the A
site. The shorter bond is also found in l
Bi1-O3
= 0.2418 nm (12% shorter)
along the b-axis and l
Bi1-O1
= 0.2527 nm (17% shorter), which are responsible
for the large TiO
6
octahedral rotation around the a-axis, as clearly seen in
Fig. 33.1(b).
It is interesting to note that a marked change in bond length is also observed
for Bi2–O. The smallest two bonds are observed in l
Bi1-O4
= 0.2556nm (12%
shorter) along the b-axis and l
Bi1-O4
= 0.2614nm (10% shorter) along the
a-axis. These bond lengths are relatively longer compared with l
Bi1-O3
. Although
the Bi2–O4 bonding contributes to the satisfactory bonding of Bi2 and to the
stabilization of the alternate stacking of the Bi
2
O
2
layers and the perovskite
layers, these bonds are suggested to play a minor role in the ferroelectricity
in the BiT system.
33.3 Electronic band structure and density of
states (DOS)
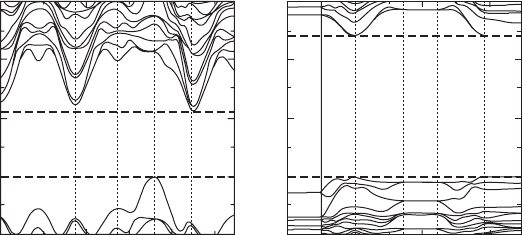
Figure 33.5 shows the electronic band structures for (a) the paraelectric
(700°C) and (b) the ferroelectric (25°C) phases of BiT calculated from the
structural data determined by the neutron diffraction study. The band dispersion
for the paraelectric phase is larger than that for the ferroelectric phase, which
is attributed to the ferroelectric displacements induced by the descending
symmetry. In the paraelectric state, the fundamental band gap (E
g
) is indicated
to be indirect (valence band maximum at P and conduction band minimum
at Brillouin-zone center Γ) [34], which is consistent with the calculation
performed by Shimakawa et al. [34]. In contrast, our calculations suggest
that the ferroelectric BiT has a direct band gap, with the valence band maximum
and conduction band minimum lying at Γ. As is typical in the calculations using
density functional theory (DFT) within generalized gradient approximation,
E
g(DFT)
is underestimated in our calculations. The experimental E
g
of 3.3eV
[35] at 25 °C is higher than the calculated E
g(DFT)
of 2.4eV. The calculated
E
g(DFT)
of the paraelectric phase is 1.1eV. The values of E
g(DFT)
agree
qualitatively with the results of the optical measurements of BiT crystals [35].
The electronic structure of the Nd-substituted BiT is discussed in Section 33.8.
WPNL2204
Handbook of dielectric, piezoelectric and ferroelectric materials1012
(a)
(b)
(c)
Ti2
Ti1
Ti2
b
c
a
Ti2
06
05
Ti1
Bi1
01
03
03
0.242
0.228
0.240
0.232
0.253
a
b
c
Bi2
Ti2
02
04
0.261
0.256
0.256
0.221
0.227
33.4
Detailed bond length in the ferroelectric state at 25 °C: (a) the crystal structure projected along the b-axis, the
enlarged views of (b) around Bi1 at the perovskite A site and (c) around Bi
2
in the Bi
2
O
2
layers.
WPNL2204
Crystal structure and defect control 1013
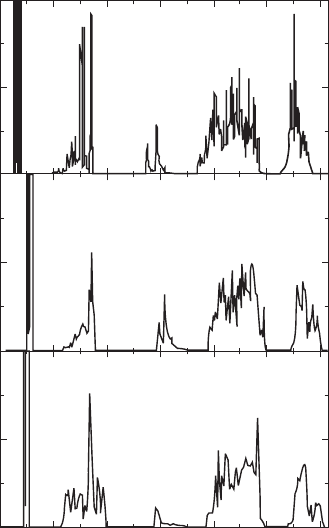
Figure 33.6 presents the total electronic density of states (DOS) between
–25 and 7.5eV, where the Fermi level (E
F
) is set at 0eV. For both the
paraelectric and ferroelectric phases, the Bi 5d states are localized at a deep
level (–22 to –24eV). The O 2s and Bi 6s stats appear at –17 to –20eV and
–7 to –12eV, respectively. The valence band consists mainly of the O2p
states, while the Ti 3d states form the conduction band. The Bi 6p states
overlap with the Ti 3d states in the conduction band.
Figure 33.7 depicts the total DOS and partial (ion decomposed and sphere
projected) DOS (PDOS) of the constituent ions of the (a) paraelectric and (b)
ferroelectric phases of BiT. The ionic model suggests that the Ti 4d states are
formed only in the conduction band, while significant PDOS of the Ti 3d
states is seen in the valence band in both the paraelectric and ferroelectric
states. The presence of the Ti 3d states in the valence band indicates that the
orbital hybridization between the Ti 3d and the O 2p occurs in the BiT
system. Although the hybridization of the Ti 3d–O 2p stabilizes the TiO
6
octahedra, this orbital interaction does not play an important role in the
ferroelectric phase transition, because the value of the Ti-3d PDOS remains
unchanged regardless the phase transition.
The PDOS analysis shows a marked change in the orbital interaction
between Bi and O induced by the transition. The primary band of the Bi 6s
appears at around –10eV, while that of the Bi 6p lies in the conduction band,
which is consistent with the ionic model. The PDOS of the O 2p is observed
to some degree in the Bi 6s band due to the hybridization of the Bi 6s–O 2p.
Furthermore, the PDOS of the O 2p is seen also in the Bi 6p band caused by
the Bi 6p–O 2p bonding and antibonding interactions. Thus, the PDOS of the
O 2p in these Bi bands can be regarded as a qualitative indicator of the
degree of the orbital hybridization between Bi–O.
3
2
1
0
–1
3
2
1
0
–1
Energy (eV)
Z
Γ
XP
Γ
N
(a) Bi
4
Ti
3
O
12
(Paraelectric,
|4|mmm
)
(b) Bi
4
Ti
3
O
12
(Ferroelectric,
B2cb
)
R
Γ
SYTZ
Γ
33.5
Band structures of Bi
4
Ti
3
O
12
calculated from the structure data
determined by the Rietveld analysis: (a) the paraelectric phase and
(b) the ferroelectric phase.
WPNL2204
Handbook of dielectric, piezoelectric and ferroelectric materials1014
In the Bi
2
O
2
layers, the Bi2 6s is strongly hybridized with the O2 2p in
both the paraelectric and ferroelectric states, and a large PDOS of the O2 2p
is seen at the same energy level in the Bi2 6s main band. The Bi1 6s–O 2p
hybridization is also recognized in both states. These results suggest that the
traditional model [36, 38, 44] based on the stereoactive lone-pair electrons
of Bi
3+
is not sufficient to explain the structural distortion in the ferroelectric
state. The phase transition leads to a marked increase in the PDOS of the O3
2p in the Bi1 6s and 6p main bands, which is consistent with the short bond
length between Bi1 and O3 in the ferroelectric state. These electronic and
structural analyses demonstrate that the hybridization of the Bi1 6s(p)–O3
2p acts as a trigger of the ferroelectric transition. This hybridization results
in a satisfactory chemical bonding of Bi1 at the A site, and then the ferroelectric
distortion is stabilized in the perovskite layers.
DOS (1/eV)
50
0
50
0
50
0
Energy (eV)
–25 –20 –15 –10 –5 0 0
(c) Bi
2
Nd
2
Ti
3
O
12
(Ferroelectric,
B2cb
)
(b) Bi
4
Ti
3
O
12
(Ferroelectric,
B2cb
)
(a) Bi
4
Ti
3
O
12
(Paraelectric,
|4|mmm
)
Bi 5
d
Bi 6
s
O 2
s
O 2
p
Ti
3
d
Bi 5
d
O 2
s
Bi 6
s
O 2
p
Ti 3
d
Ti 3
d
O 2
p
Bi 6
s
O 2
s
Bi 5
d
Nd 4
p
33.6
Total electronic density of states (DOS) of (a) the paraelectric
phase of Bi
4
Ti
3
O
12
, (b) the ferroelectric phase of Bi
4
Ti
3
O
12
, and (c) the
ferroelectric phase of Bi
2
Nd
2
Ti
3
O
12
. In the calculations, the perovskite
A site is assumed to be occupied only by Nd.
WPNL2204
Crystal structure and defect control 1015
33.4 Defect structure
33.4.1 Neutron diffraction study
The information of oxygen vacancies in BiT has been obtained by the Rietveld
analysis of the time-of-flight neutron powder diffraction data measured at
700°C in vacuum [19]. The refined crystal structure with the thermal ellipsoids
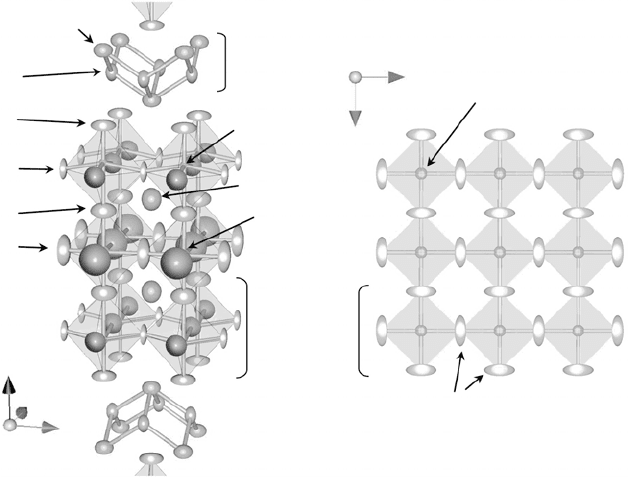
(probability 80%) is depicted in Fig. 33.8. It is interesting to note that the
thermal ellipsoid of O1 is large compared with those of other oxide ions and
that the nuclear density of O1 spreads in the a–a plane. This result strongly
indicates that self-diffusion of oxide ions occurs through the O1 site at which
a large number of oxygen vacancies
(V )
O
••
are present. Takahashi et al. [17]
have demonstrated, through the high-temperature impedance spectroscopy
study, that oxide ion is the dominant carrier for high electrical conduction
along the a-axis in BiT crystals [19], which is consistent with the results of
this neutron structural study.
33.4.2
Ab initio
band structure calculations
Vacancy formation energies of Bi and O in the paraelectric state in the BiT
system obtained by ab initio band structure calculations have been reported
Energy (eV)
–12 –8 –4 0 4
–12 –8 –4 0 4 –12 –8 –4 0 4
DOS (1/eV)
2eV
0.5 eV
20eV
Total
Total
Total
(c) Bi
2
Nd
2
Ti
3
O
12
ferroelectric phase
(
B2cb
)
(b) Bi
4
Ti
3
O
12
ferroelectric phase
(
B2cb
)
(a) Bi
4
Ti
3
O
12
paraelectric phase
(
|4|mmm
)
6
p
5
d
6
s
5
p
O6 2
p
O5 2
p
O4 2
p
O3 2
p
O2 2
p
O1 2
p
Ti2 3
d
Ti1 3
d
Bi2
Nd
6
p
6
p
6
s
6
s
Bi2
Bi1
6
p
6
p
6
s
6
s
Bi2
Bi1
O6 2
p
O5 2
p
O4 2
p
O3 2
p
O2 2
p
O1 2
p
Ti2 3
d
Ti1 3
d
O5 2
p
O4 2
p
O3 2
p
O2 2
p
O1 2
p
Ti2 3
d
Ti1 3
d
33.7
Partial (ion decomposed and sphere projected) DOS (PDOS) of
(a) the paraelectric phase of Bi
4
Ti
3
O
12
, (b) the ferroelectric phase of
Bi
4
Ti
3
O
12
, and (c) the ferroelectric phase of Bi
2
Nd
2
Ti
3
O
12
. In the calculations,
the perovskite A site is assumed to be occupied only by Nd.
WPNL2204
Handbook of dielectric, piezoelectric and ferroelectric materials1016
[19]. The supercell method is adapted for estimating vacancy formation
energy (E
vacancy
) to reduce vacancy–vacancy interaction. Figure 33.9 indicates
the relative E
vacancy
of Bi and O. Since Bi1 and O2 show the lowest E
vacancy
in Bi and O, these values are set to be the standard for comparison. The
calculations for BiT show that the E
vacancy
of Bi1 is lower by about 2eV than
that of Bi2. The much lower E
vacancy
of Bi1 indicates that Bi vacancies are
created preferentially at the Bi1 site in the perovskite layers rather than at the
Bi2 site in the Bi
2
O
2
layers. For oxygen vacancies, O1, O2 and O3 show
similar E
vacancy
within 0.2 eV, which is lower by about 0.4–0.6 eV than those
of O4 and O5. It is concluded that Bi1 vacancies and O1 vacancies are
primary ionic defects in BiT at high temperatures.
It is interesting to note that the presence of La at the perovskite A site
increases E
vacancy
of the adjacent oxygen atoms, O1, O3 and O5, by about
1eV, as shown in Fig. 33.9. These results suggest that La substitution is
effective for improving the chemical stability of oxygen in the perovskite
layers. The total/partial DOS analysis of BLT (x = 2.0) suggests that the La
5d is hybridized with the O 2p of the adjacent oxygen. The covalent bonding
between La and O in the perovskite layers creates the DOS of the La 5d
states in the valence band near the Fermi level (–5~0eV), which is a possible
origin of the high chemical stability of oxygen in BLT. The chemical
(b)
(a)
TiO
6
octahedron
O1
Ti1
a
a
a
a
c
Ti1
Bi1
Ti2
Bi
2
O
2
layer
Bi2
O2
O4
O5
O3
O1
33.8
Refined crystal structure with thermal ellipsoids (probability
80%) of BiT at 700°C by neutron diffraction Rietveld analysis.
WPNL2204
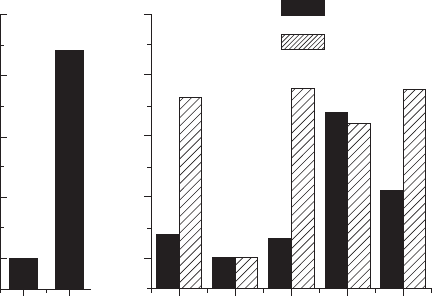
Crystal structure and defect control 1017
stability of oxide ions seen in BLT is confirmed also in BNT by ab initio
calculations.
33.4.3 Electrical conductivity investigations by ac
impedance spectroscopy
Figure 33.10 shows the temperature dependence of electrical conductivity
(σ). The σ along the a-axis of BiT was about two orders of magnitude higher
than that along the c-axis. Takahashi et al. have revealed that the high σ
along the a axis is attributed to oxide ion conduction in the perovskite layers
[17, 18]. In contrast, the Bi
2
O
2
layers act as a barrier to oxide ion migration
along the c-axis; hence this yields the low σ along the c-axis (h
•
conduction
is dominant). The oxide ion migration is achieved through O1 vacancies, and
oxide ions are suggested to move in the middle of the perovskite layers in the
a–a (a–b) plane.
The σ along the c-axis of BLT (x = 0.46) crystals was the same as that of
BiT, while the La substitution decreases σ along the a-axis. In BLT (x =
0.46), most La occupies the perovskite A site [33]. The same σ along the
c-axis for BiT and BLT suggests that the Bi
2
O
2
layers play the same role for
BiT and BLT in the electrical conduction along the c-axis. The lower σ along
the a-axis of BLT strongly indicates that the La substitution is effective for
decreasing
(V )
O1
••
, which is essentially consistent with the results of the
E
vacancy
calculations. The relatively high oxide-ion conductivity observed for
BLT (x = 0.46) indicates that a certain amount of
(V )
O1
••
is present even in the
lattice.
Bi
4
Ti
3
O
12
Bi
2
La
2
Ti
3
O
12
Relative
E
vacancy
(eV)
2.0
1.5
1.0
0.5
0.0
2.0
1.5
1.0
0.5
0.0
Bi1 Bi2
O1 O2 O3 O4 O5
33.9
Relative difference in vacancy formation energy (
E
vacancy
) in
I4/mmm
tetragonal symmetry calculated by ab initio band structure
calculations. In the calculations for BLT, the perovskite A site is
assumed to be occupied by La.
WPNL2204
Handbook of dielectric, piezoelectric and ferroelectric materials1018
33.4.4 Defect-formation mechanism
Here, a mechanism of vacancy formation in BiT at high temperatures is
discussed. It has been shown that the Bi vacancy at the Bi1 site
(V )
Bi1
′′′
is
more easily generated than
′′′
V
Bi2
, where ′ denotes one negative charge (Fig.
33.9). Furthermore, the nuclear-density analysis has revealed that oxygen
vacancies preferentially reside at the O1 site
(V )
O1
••
, where
•
indicates one
positive charge (Fig. 33.8). The vacancy formation of
′′′
V
Bi1
is likely to be the
trigger for generating
V
O1
••
. At high temperatures,
′′′
V
Bi1
is formed accompanied
with the nearest-neighbor
V
O1
••
, which is the origin of the defect formation in
the BiT system. The high oxide-ion conduction observed in the BiT crystals
is ascribed to the large number of
V
O1
••
at the perovskite layers. The high
oxide-ion conductivity also indicates that the electrical neutrality during the
defect formation is satisfied through the generation of
2V
Bi1
′′′
accompanied
with
3V
O1
••
as follows:
2Bi + 3O 2V + 3V + Bi O (g)
Bi1
O1
Bi1
O1 2 3
→
′′′
••
33.2
and the electron hole (h
•
) plays a minor role in the high-temperature defect
formation.
As a result of the thermogravimetric analysis of BiT powder under different
oxygen partial pressures
()
O
2
P
[19], the weight loss due to the vacancy
formation was promoted in low-
P
O
2
(reducing) atmosphere. The weight loss
T
–1
(10
3
K
–1
)
1.31.21.11.0
log (σ/S cm
–1
)
–2
–1
–3
–4
–5
–6
In air
BiT
a
-axis
BLT
(
x
= 0.46)
c
-axis
33.10
Electrical conductivity in air as a function of temperature
observed for the crystals of BiT and Bi
4–
x
La
x
Ti
3
O
12
(BLT,
x
= 0.46).
WPNL2204
Crystal structure and defect control 1019
due to the vacancy formation was observed above 1000°C in air [19]. Annealing
in air at 1000°C for 5 h led to a 0.25% decrease in weight, while annealing
in O
2
( = 0.1 MPa)
O
2
P
at 1000 °C did not result in weight loss. The O
2
annealing at 1050°C for 5 h resulted in a weight loss of 0.5%, which was
much smaller than the annealing at 1050°C for 5h in air (1.2% loss). The
P
O
2
dependence of weight loss shows that the limiting factor of the vacancy
formation is the surface reaction on BiT powder, and this result gives valuable
information that the vacancy formation can be suppressed by heat treatment
in high-
P
O
2
atmosphere [19].
Thermogravimetric analysis allows us to propose the mechanism of reaction
on the BiT surface that leads to the vacancy formation from the three possible
models. In model 1, two lattice Bi1
(Bi )
Bil
x
and three lattice O1
(O )
O1
x
migrate
to the surface and react with each other to form Bi
2
O
3
. The resultant Bi
2
O
3
evaporates into the atmosphere, i.e. Bi
2
O
3
(g). This reaction, however, should
be independent of
P
O
2
and thus does not explain the experimental results. In
model 2, only
Bi
Bil
x
moves from the lattice to the surface. Then, the adsorbed
Bi (Bi
ad
) reacts with O
2
gas in the atmosphere, O
2
(g), and evaporates as
Bi
2
O
3
(g). Although this reaction yields
′′′
V
Bi1
and electron holes (h
•
), an
increase in
P
O
2
should enhance the weight loss, i.e.
′′′
V
Bi1
formation, which
is not consistent with the experiments. In model 3, two
Bi
Bil
x
and three
O
O1
x
migrate to the surface, but do not react with each other because of the low
possibility of Bi
ad
meeting O
ad
on the surface (due to low vacancy
concentrations), as schematically depicted in Fig. 33.11. Bi
ad
reacts with
O
2
(g) and evaporates as Bi
2
O
3
(g) at a relatively higher
P
O
2
, while Bi
ad
detaches from the surface as Bi(g) or Bi clusters in reducing atmosphere.
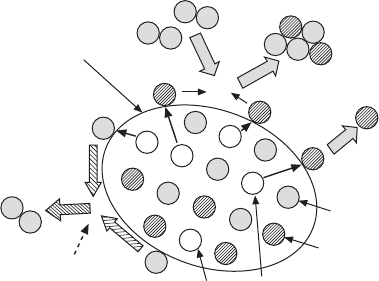
Rate-limiting
reaction
O
ad
V
O1
Bi
Bi1
″′
. .
Bi
Bi1
x
O
O1
x
Bi
ad
Bi (g)
Bi
2
O
3
(g)
O
2
(g)
O
2
(g)
O
ad
BiT surface
33.11
Mechanism of vacancy formation in BiT at high temperatures.
Bi
Bi1
x
is Bi at the Bi1 site with no charge and
O
O1
x
denotes O at the
O1 site with no charge.
Bi
Bi1
x
and
O
O1
x
migrate to the surface and
then become adsorbed Bi
ad
and O
ad
. The limiting factor of the
vacancy formation is the surface reaction related to O
ad
.
WPNL2204