Czichos H., Saito T., Smith L.E. (Eds.) Handbook of Metrology and Testing
Подождите немного. Документ загружается.


258 Part B Chemical and Microstructural Analysis
0.1 mm
a) b)
Fig. 5.79a,b Dislocation contrasts observed in an indented CdTe
surface. (a) SEM-CL image showing dislocations in dark contrast
and (b) OM image showing dislocation etch pits developed after
(a) was recorded
waves resulting in diffraction peak broadening and
peak shifts along the direction normal to the defect
planes. Analogously to the effect of finite crystal size
on diffraction, the peak broadening width is inversely
proportional to the mean distance between adjacent
fault planes. Unlike the size effect, however, the ef-
fects are different depending on the nature of the
defects. The peak broadening is symmetric in stack-
ing faults but asymmetric in twin boundaries. The peak
shift is absent for some types of stacking faults but
is induced in directions that depend on the types of
stacking faults in other cases. The analytical detail
may be found in a comprehensive textbook by Snyder
et al. [5.72].
Mechanical Spectroscopy
Internal friction (Sects. 5.1.2 and 5.3.1) provides a sen-
sitive tool to study the motion of dislocations at very
low stresses. A typical application is to elementary
processes of dislocation motion in metals and ionic
crystals. Dislocation pinning is applied to detect the
migration of defects and impurities.
Recoverable atomic motion in grain boundaries
can be studied by means of mechanical spectroscopy
(Sect. 5.1.2), e.g., the anelastic relaxation of grain
boundaries is observed above room temperature in
coarse-grained fcc metals but below room temperature
in nanocrystalline fcc metals.
5.4 Molecular Architecture Analysis
The materials in the scope of this section include
simple molecules, polymers, macromolecules (super-
molecules) as well as biomolecules such as proteins.
The rapidly advancing techniques relating to DNA anal-
ysis are out of the scope of this section. Molecules
on large scales can have higher-order structures that
perform important functions, especially in biopoly-
mers. The primary structure of proteins, for example, is
a sequence of amino acids (peptide) that constitutes sec-
ondary structures in forms such as helices and sheets.
The secondary structures linked in a protein are usually
folded to tertiary structures that may further become as-
sociated to a quaternal structure that brings about such
bioactivity as allosteric effects. This section addresses
mainly nuclear magnetic resonance (NMR) in consid-
erable detail for its unique power in the analysis of the
architecture of macromolecules. Although single crys-
tal x-ray diffraction is another important technique for
structural determination of macromolecules, we only
briefly mention some points specific to macromolecu-
lar samples. The large molecules may be pretreated by
chromatographic techniques to separate or decompose
them into constituents or smaller fragments that can
be analyzed by simpler methods. After the preprocess-
ing, the molecules or the fragments may be subjected
to standard analysis such as FT-IR, Raman scatter-
ing, and fluorescence spectroscopy for identification of
the constituent bases, as described in other sections
(Sects. 5.1.2 and 5.2.3). In this section, we mention
only optical techniques based on the circular dichroism
exhibited by helix molecules and the fluorescence reso-
nant energy transfer (FRET) that provides information
on the proximity of two stained molecules.
5.4.1 Structural Determination
by X-Ray Diffraction
The principle of structural analysis by x-ray diffraction
in macromolecules is essentially the same as that of sin-
gle crystal diffraction and powder diffraction already
described in Sect. 5.1.1. For the growth of molecu-
lar crystals, the synthesis of the material molecules is
necessary. Thanks to progress in genetic engineering
such as the advent of the polymerase chain reaction
(PCR) technique, even large biomolecules such as pro-
teins can be synthesized in sufficient amounts. However,
macromolecules and polymers tends to be condensed to
amorphous or very disordered aggregates (Sect. 5.2.2);
Part B 5.4

Nanoscopic Architecture and Microstructure 5.4 Molecular Architecture Analysis 259
the growth of single crystals at high quality is gener-
ally difficult for such materials. The direct method is
applicable only to molecules of 100 or fewer atoms
because, as molecular size increases, the ambiguity in
phase determination rapidly increases. The heavy atoms
substitution method works for molecules larger than
600 atoms. So a gap is present for molecules in the inter-
mediate size range. In principle, as long as a good single
crystal sample is obtained, there is no limitation on the
molecular size. The maximum size so far achieved by
making full use of up-to-date methods is as large as
that of ribosome (4 × 10
6
in molecular weight), though
normally, the maximum size subject to routine analysis
is ≈10
4
in molecular weight. The powder diffraction
technique is also applicable to large molecules, but the
accuracy is limited due to the difficulty in separation of
diffraction peaks, which are more crowded than from
small molecules. The maximum molecular size is sev-
eral thousands of atoms. For more details on x-ray
structural analysis of macromolecules, see [5.73].
5.4.2 Nuclear Magnetic Resonance (NMR)
Analysis
Standard structural analysis by NMR proceeds follow-
ing (1) sample preparation, (2) NMR spectra measure-
ments, (3) spectral analysis to assign NMR signals to
the responsible nuclei and find the connectivity of the
nuclei through bonds and space, (conventionally the
term signals is used rather than peaks because NMR
resonances are not always observed as peaks.) and fi-
nally (4) the deduction of structural models using the
knowledge obtained in (3) as well as information from
other chemical analyses as a constraint in the proce-
dure of fitting model to experiment. The final step (4)
is like forming a chain of metal rings of various shapes
(e.g., amino acid residues in proteins) on a frame hav-
ing knots in some places linked to others on the chain.
For this reason, particularly for macromolecules such as
proteins, it is difficult to determine the complete mo-
lecular structure uniquely from NMR analysis alone.
Since we can only deduce possible candidates for the
structure, it is better to refer to models rather than struc-
tures. This is in marked contrast to structural analysis by
x-ray diffraction in which the crystal structure is more
or less determined from the diffraction data alone. Nev-
ertheless, NMR has advantages over x-ray diffraction
methods in many respects such as
1. Single crystal samples are not needed. The samples
may be amorphous or in solution.
2. Effects of intermolecular interactions, that may
change the molecular structure, can be avoided by
dispersing samples in suspending solution.
3. Dynamic motion of molecules can be detected.
4. A local structure can be selectively investigated
without knowing the whole structure.
5. Fatal damage due to intense x-ray irradiation, that
is likely to happen in organic molecules, can be
avoided.
Points 2–4 are of particular importance for proteins
that function in solution, changing their local conforma-
tional structure dynamically.
In this chapter, we will describe only steps (2) and
(3) in some detail, leaving (1) and (4) to good text-
books [5.74–76] except for a few words on step (1).
Before proceeding to the experimental details, we
briefly summarize the information that NMR spectra
contain.
Information Given by NMR Spectra
Nuclei other than those containing both an even num-
ber of protons and neutrons (such as
12
Cand
16
O)
have a nonzero nuclear spin I. The magnetogyric ra-
tio γ
n
giving the nuclear magnetic moment (Sect.5.1.2)
is a natural constant specific to the nuclear species.
Table 5.4 lists isotopic nuclei commonly contained in
organic molecules having hydrocarbons in the back-
bones. Among them, proton
1
H, carbon
13
Cand
nitrogen
15
N are characterized by the smallest nuclear
spin 1/2. As explained later, this fact, together with the
fact that
1
H in very high natural abundance has a large
γ
n
value, provides the reason why mainly these isotopes
are used for high-resolution NMR measurements. As far
as stated in the following, we consider only the case of
I = 1/2 for simplicity. The values of the Larmor (not
angular) frequency at a typical magnetic field of 2.35 T
are listed in the fifth column in Table 5.4 for various iso-
lated nuclei. It should be noted that, with increasing γ
n
,
the energy difference of the two spin states, and hence
the population difference and the magnetization to be
detected in experiments, increase, so the sensitivity of
NMR is high particularly for protons that have a large
γ
n
value.
The magnetic field B
0
felt by the nuclear spins may
differ from the external field due to many causes.
Chemical Shift. The external magnetic field is shielded
by the diamagnetic current of s electrons or enhanced
by the paramagnetic current of p and d electrons sur-
rounding the nucleus. In contrast to the shielding field
Part B 5.4
260 Part B Chemical and Microstructural Analysis
Table 5.4 Properties of nuclear species commonly used for NMR analysis of macromolecules. After [5.77]
Nuclear species Nuclear spin I Magnetogyric ratio Natural abundance Larmor frequency
() γ
n
(10
8
rad s
−1
T
−1
) (%) (MHz)
a
1
H 1/2 18.861 99.985 100.000
2
H 1 2.895 0.015 15.351
3
H 1/2 20.118 Radioactive 106.663
12
C 0 − 98.0 −
13
C 1/2 4.743 1.1 25.144
14
N 1 1.363 99.634 7.224
15
N 1/2 −1.912 0.366 10.133
16
O 0 − 99.762 −
17
O 5/2 −2.558 0.038 13.557
18
O 0 − 0.200 −
a
NMR resonance frequency of isolated nuclei at a typical magnetic field of 2.35 T
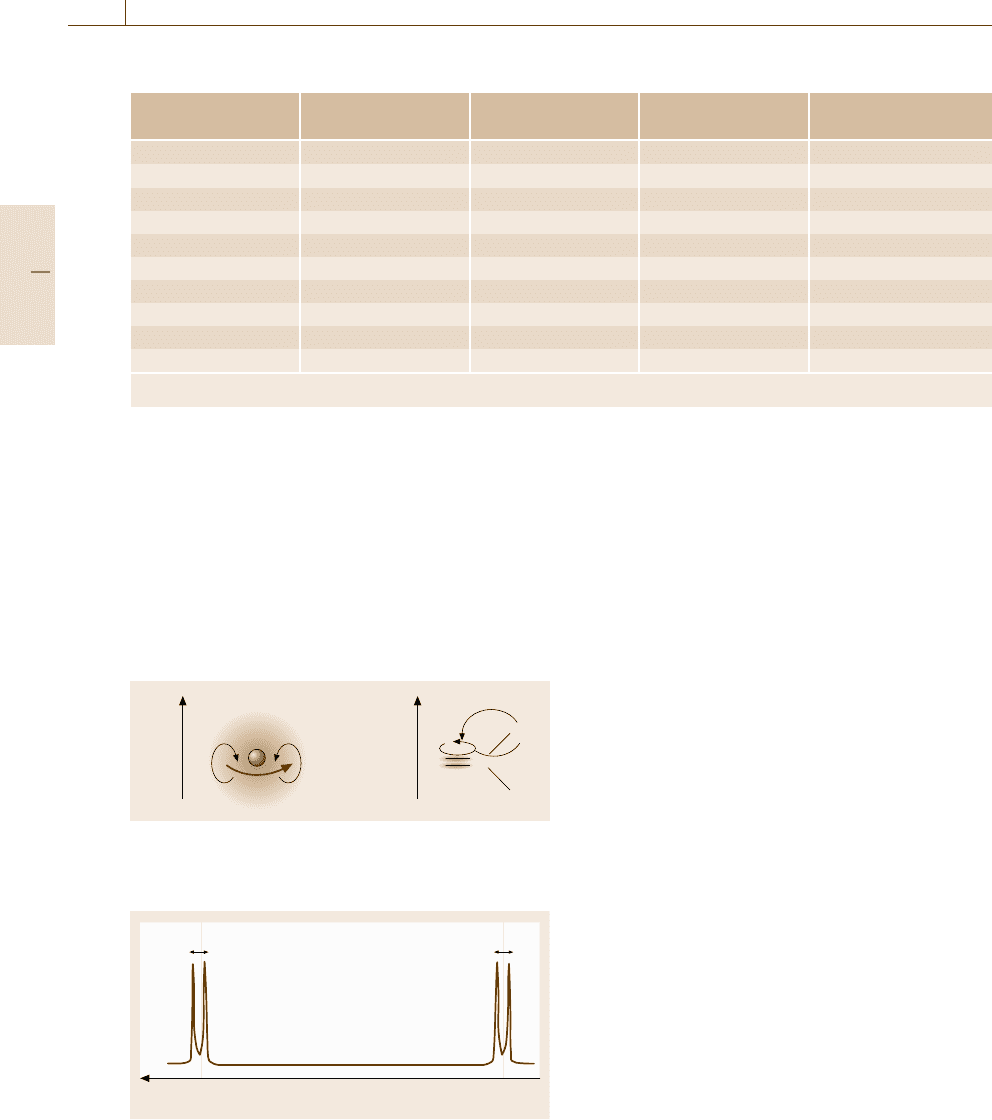
from s electrons (Fig. 5.80a), which is isotropic in the
sense that it is directed along the magnetic field due to
the isotropic nature of s electrons, the shielding field
is anisotropic when the electrons are, e.g., π electrons
in double bonds (Fig. 5.80b). In all these cases, the
applied magnetic field induces a screening current of
electrons surrounding the nucleus which exerts an ad-
ditional local field on that nucleus. Since the degree
of screening depends on the chemical environment of
the nucleus, the difference in the nuclear environment
B
0
σB
0
e
–1
B
0
C
H
H
σB
0
e
–1
a) b)
Fig. 5.80a,b The magnetic shielding by electrons induc-
ing the chemical shifts in NMR. (a) Isotropic shielding by
s electrons, (b) anisotropic shielding by π electrons
H2 H1
J
H1-H2
(Hz) J
H1-H2
(Hz)
1
H (ppm)
Fig. 5.81 Chemical shifts and J-coupling in molecules
containing two protons in different environments (sche-
matic)
results in a small shift, called the chemical shift δ,of
the resonance frequency. As illustrated by a schematic
NMR spectrum in Fig. 5.81 of protons in a molecule
which contains two protons at different positions, the
chemical shift can differ depending on the chemical
environment. Thus, the chemical shift is used to dis-
criminate the position in a molecule at which the probed
nuclei are situated. Since the chemical shift increases
proportionally with the external field, the shift is usually
expressed by a fraction (in unit of ppm) of the Larmor
frequency of the nucleus, which also increases linearly
with the field.
Spin–Spin Coupling (Connection Through Bonds). In
Fig. 5.81, the two sets of spectral signals corresponding
to different chemical shifts further splits to two sig-
nals with small separations. This is due to the spin–spin
coupling mediated by electrons contributing chemical
bonds linking the two spins: as shown in the top left
illustration in Fig. 5.82, a nuclear spin interacts with
electrons surrounding the nucleus, then the spins of the
electrons interact with other electrons through an ex-
change interaction when chemical bonds are formed,
and the latter electrons interact with another nuclear
spin. Unlike the direct dipole–dipole interaction be-
tween magnetic moments that operates only in a small
distance, such indirect spin–spin coupling extends to
a larger distance of up to 3–4 bonds. In contrast to the
chemical shift, the magnitude of the spin–spin coupling
is independent of the external magnetic field and there-
fore the splits of the resonance frequency due to the
spin–spin coupling are expressed by a coupling con-
stant J scaled in units of microwave frequency. Due
to the origin of the spin–spin coupling (hereafter called
Part B 5.4
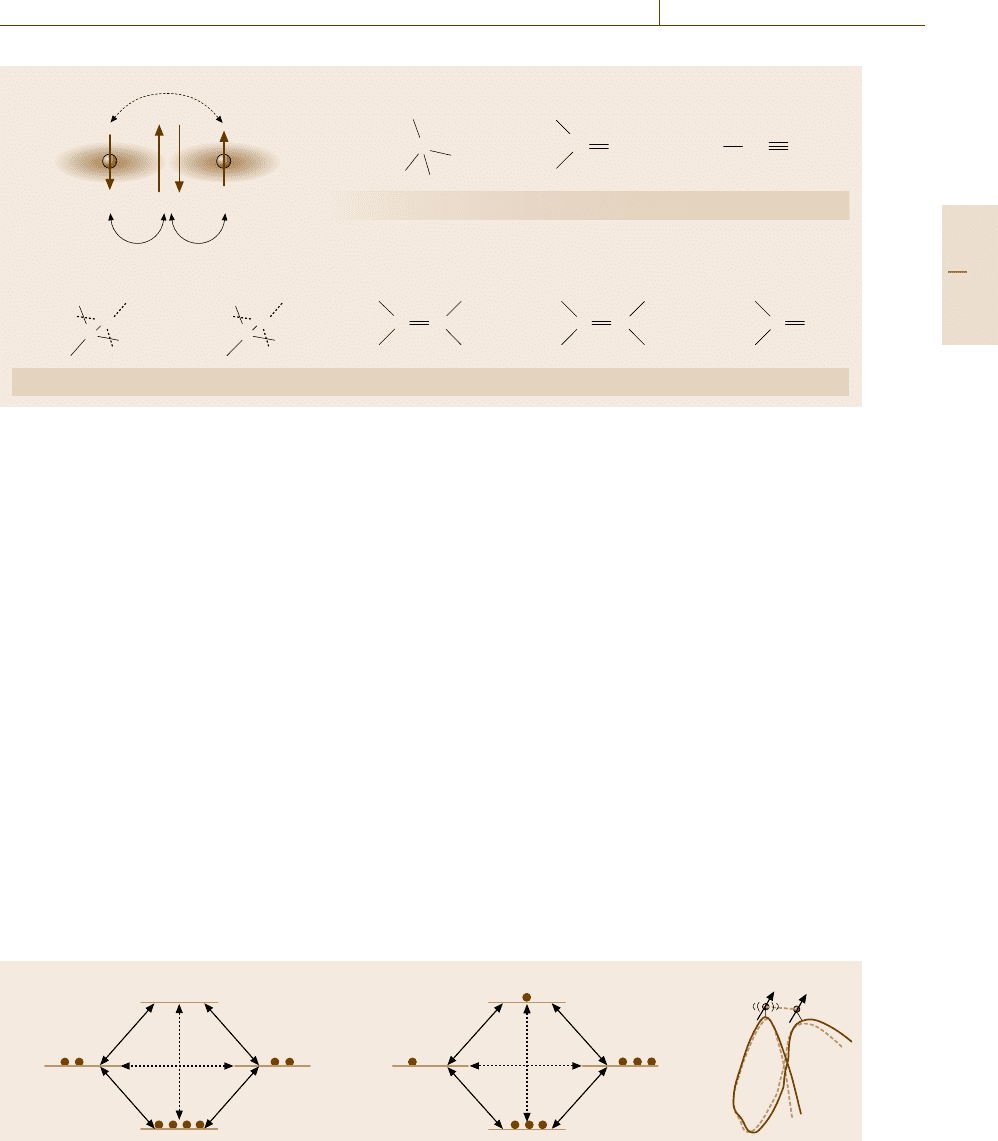
Nanoscopic Architecture and Microstructure 5.4 Molecular Architecture Analysis 261
CC
Xe
–1
A
J
13
C
13
C
1
H
1
H
1
H
sp
3
sp
2
sp
J = 120–150Hz 150–170 Hz 240–260 Hz
J =6–12 Hz 2–5 Hz 12–19Hz 5–11 Hz –3–2 Hz
trans gauche trans cis gem
C
1
H
13
C
C
1
H
C
1
H
C
1
H
1
H
1
H
1
H
CC
1
H
1
H
C
1
H
Fig. 5.82 J-coupling in various bases
J-coupling), the value of J can vary and even change
its sign depending on the character and the number
of chemical bonds intervening between the two spins.
Some typical values of J are indicated in Fig. 5.82 be-
tween two
1
H nuclei or
1
Hand
13
C connected with
different types of chemical bonds. Thus, the value of
the spin coupling constant J reflecting the interaction
through bonds provides information of the steric local
configuration of the molecule.
Nuclear Overhauser Effect (Connection Through
Space). In contrast to the spin–spin interaction me-
diated by electrons, the magnetic moments associated
with two nuclei can directly interact through the clas-
sical dipole–dipole interaction. Since the strength of
the dipole–dipole interaction depends quadratically
on the product of the magnetogyric ratios and de-
cays rapidly with the internuclear distance d as d
−3
,
the dipole–dipole interaction acts virtually only be-
tween two proton nuclei within a distance of ≈ 5Å
in diamagnetic molecules. Since electrons inducing
paramagnetism have a large magnetogyric ratio, they in-
β
I
α
S
a) b) c)
β
I
α
S
α
I
β
S
α
I
β
S
μ
I
μ
S
d
β
I
β
S
α
I
α
S
W
1I
W
1I
W
2
W
0
α
I
α
S
W
2
W
0
W
1S
W
1S
β
I
β
S
W
1I
W
1I
W
1S
W
1S
Fig. 5.83a–c Level diagrams illustrating the nuclear Overhauser effect (NOE), which operates between nuclei close in
space. For details, see the text
teract so strongly with nuclear spins that NMR becomes
very broad or unobservable.
The dipole–dipole interaction gives rise to the fol-
lowing nuclear Overhauser effect (NOE) that provides
powerful methods for structural determination. Gener-
ally, systems of nuclear spins, once disturbed, attempt
to recover their equilibrium state. The recovery or re-
laxation takes place via two different processes: the
spin–lattice relaxation in which the excited energy is re-
leased to the heat bath (usually lattice) by flipping of
spins, and the spin–spin relaxation in which only the
coherence of spins is lost without energy dissipation.
The relaxation processes are characterized by first-order
time constants, called the vertical time constant T
1
for
the former and the transverse time constant T
2
for the
latter.
As an illustrative relevant example, we consider two
protons, I and S, that are supposed not to be J-coupled,
for simplicity. The energy level diagram of the two-spin
system in thermal equilibrium is shown in Fig. 5.83a,
in which the state α
I
β
S
, for example, indicates that the
spin of I is up (α) while the spin of S is down (β). In
Part B 5.4

262 Part B Chemical and Microstructural Analysis
the absence of J-coupling, the levels α
I
β
S
and β
I
α
S
are
nearly degenerate with a small difference due to pos-
sible chemical shifts in I and S. The number of dots
on each level indicates the level population. Now, we
assume that, as shown in Fig. 5.83b, the S spins are se-
lectively irradiated with microwaves at the resonance
frequency so that the populations are equalized between
the levels linked by W
1S
, called single-quantum pro-
cesses, achieved by the flipping of a single spin. Once
the populations are disturbed this way, spin–lattice re-
laxation occurs via single-quantum transitions between
α
I
α
S
and β
I
α
S
, α
I
β
S
and β
I
β
S
for I, and between α
I
α
S
and α
I
β
S
, β
I
α
S
and β
I
β
S
for S, both observable as
singlet NMR spectra. The spin–lattice relaxation may
also occur by transitions between α
I
α
S
and β
I
β
S
, called
double-quantum processes W
2
, that correspond to si-
multaneous flipping of both spins. The populations may
also be recovered by the transition between β
I
α
S
and
α
I
β
S
, called a zero-quantum process W
0
, which causes
spin–spin relaxation. The relaxation through W
0
and
W
2
are normally forbidden but becomes allowed when
there are magnetic field fluctuations that act as an alter-
nating field at various frequencies, which induces spin
flipping. The field fluctuations are generated by the dif-
fusional or rotational motion of nuclei and molecules
(Fig. 5.83c) that cause fluctuations in the local field af-
fected by the dipole–dipole interaction. Generally, the
fluctuation-induced relaxation rate becomes maximum
when the resonance condition ω
0
τ
c
≈ 1 is satisfied,
where τ
c
is the correlation time of the fluctuation and
ω
0
is the relevant transition energy. Since ω
0
=ω
L
for
W
1
and ω
0
= 2ω
L
for W
2
, normally ω
0
τ
c
1inlarge
molecules or molecules in viscous solution for which τ
c
is large, and hence relaxation through W
1
and W
2
is in-
efficient in such systems. For the process W
0
,however,
the two levels are close in energy (ω
0
≈0) so that re-
laxation through W
0
can be efficient when ω
0
τ
c
≈1is
satisfied, thereby dominating the spin–spin relaxation.
In any cases, the excited spin systems recover their
thermal populations with a rate affected by the cross re-
laxation processes W
0
and W
2
due to the dipole–dipole
interaction, if present. This NOE effect, originating
from an interaction through space, is exploited for de-
termination of the conformation of macromolecules.
Experimental Methods
Vector Model in Rotating Frame. In modern NMR ex-
periments, we do not anymore measure the absorption
of a continuous microwave, but instead measure a re-
sponse of the spin system to a sequence of microwave
pulses and Fourier transform the oscillatory current sig-
nal response to the pulses to obtain an NMR spectrum.
Experimentally, in addition to the static magnetic field
along the z-axis, we apply another magnetic field to the
spins with a small intensity 2B
1
alternating at an angu-
lar frequency ω with a coil (transmitter) that is wound
around the sample with its axis (taken as x-axis) nor-
mal to the z-axis. One of the circularly rotating fields of
intensity B
1
gives rise to two effects: it forces the pre-
cession of the spins to synchronize and to tip away from
the z-axis. A simple calculation shows that, if one views
the precessing spin from the coordinate system O-x
y
z
rotating around the z-axis at the alternating frequency ω,
the spin feels an effective magnetic field composed of
a static field B
1
directing along the x
-axis and a con-
stant field B
0
−ω/γ
n
along the z-axis. Therefore, if we
prepare spins under a static field B
0
and at time t =0we
apply a rotating field B
1
that satisfies the resonance con-
dition ω =ω
L
(=γ
n
B
0
), only the field B
1
remains and
causes the spins to turn coherently around the x
-axis at
a small angular frequency of γ
n
B
1
. Therefore, if we fo-
cus on the magnetization, the average of the magnetic
moments associated with spins rather than the spins
themselves, we can draw pictures like Fig. 5.36a–c, in
which the magnetization is represented by a thick vec-
tor. Hereafter we will repeatedly use such classical vec-
tor models in rotating frames for intuitive interpretation.
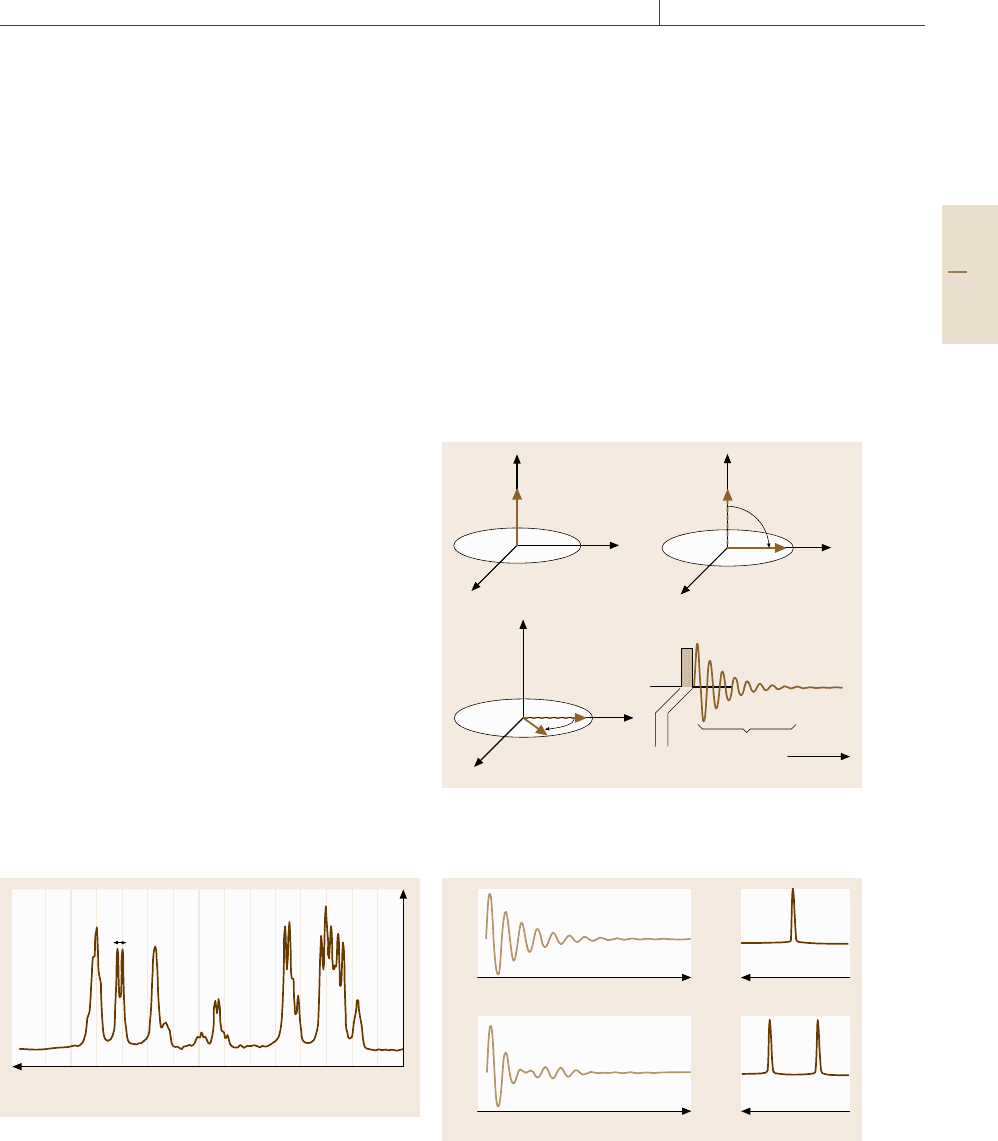
High-Resolution NMR. As mentioned already, the ab-
solute magnitude of chemical shifts becomes larger as
the external magnetic field B
0
is increased while the
fine structure due to J-coupling is fixed in magnitude.
Therefore, the use of a higher magnetic field allows one
to resolve overlapping signals closely split with very
small differences in chemical shifts (Fig. 5.84). Espe-
J
2
J
1
Higher B
0
ΔδαB
0
Fig. 5.84 Chemical shifts Δδ increasing with magnetic
field and invariable spin–spin couplings J
1
and J
2
. The use
of high field enhances the resolution of NMR
Part B 5.4
Nanoscopic Architecture and Microstructure 5.4 Molecular Architecture Analysis 263
cially in macromolecules such as proteins, substantial
overlapping of NMR spectra are common and the re-
moval of the overlaps is requisite. One experimental
solution is to use an extremely high-field (superconduct-
ing) magnet the strength of which being, in terms of
the equivalent
1
H resonance frequency, as high as, e.g.,
800 MHz.
One-Dimensional (1-D) NMR. Free Induction De-
cay (FID). An extensive range of modern techniques
developed for NMR experiments are solely devoted
to separate complicated signals, such as shown in
Fig. 5.85, without artifacts. Some of the tasks can be
solved, as well as through the use of high-field magnets,
by one-dimensional NMR (1-D-NMR) schemes that are
illustrated by pulse sequence diagrams. The simplest
case, though rarely encountered in practice, is illustrated
by a vector model shown in Fig. 5.86a–c. In the ini-
tial state (Fig. 5.86a) the magnetization vector is aligned
along the z-axis, and in the second step (Fig. 5.86b) we
apply a rotating field at a reference frequency ω for
a duration Δt that just satisfies γ
n
B
1
Δt = π/2sothat
the magnetization is tipped onto the y
axis. We may
not be able a priori to set the reference frequency ω
identical to the resonance Larmor frequency ω
r
to be
measured. In such off-resonance cases, the magnetiza-
tion vector in the rotating frame would rotate with a rate
ω
r
−ω. In the third step (Fig. 5.86c), since the rotating
field is switched off (free), the coherent magnetization
remains in the y
axis. This means that the magneti-
zation starts to rotate in the laboratory frame with the
Larmor frequency ω
r
and can be detected as an oscilla-
tory induction current flowing the transmitter coil, now
acting as a receiver coil, as illustrated in Fig. 5.86d, with
a decay due to relaxation processes. When the nuclei are
isolated such that they have a single Larmor frequency,
1
H (ppm)
7.77.87.98.08.18.28.38.48.58.68.78.88.9 7.6
J
NH-H
α
Fig. 5.85 NMR spectrum showing chemical shifts and
J-coupling in cyclic-GRGDSPA peptides (Courtesy of
Prof. I. Shimada)
the Fourier transform of the free induction decay (FID)
curve has a signal at the Larmor frequency, as shown in
Fig. 5.87a, which represents the NMR spectrum of the
nuclei.
When multiple resonance signals coexist, the du-
ration of the π/2 pulse Δt should be short enough
(≈10 μs, with B
1
being correspondingly large) for the
alternating frequency to spread over a range covering
the chemical shifts of the nuclei concerned, so that all
the nuclei can be tipped nonselectively with a single
pulse. Such pulses are called hard pulses. In contrary,
the longer and weak pulses, called soft pulses, tip a spe-
cific nucleus selectively. The FID and NMR spectrum
in Fig. 5.87b are those obtained by using a hard π/2
pulse. Conventionally NMR spectra are displayed with
the frequency axis increasing to the left.
a) b)
c) d)
z
M
x'
y'
z
x'
π/2 pulse
π/2 pulse
Time
FID
a) b) c)
y'
z
x'
y'
Fig. 5.86 (a–c) The magnetization response in vector
model in the rotation frame and (d) free induction decay
(FID)
Frequency
a)
Time
b)
Time
Frequency
Fig. 5.87a,b Fourier transform NMR spectra (right) ob-
tained from FID curves (left)
Part B 5.4
264 Part B Chemical and Microstructural Analysis
More general procedures of 1-D-NMR consist of
first to perturb the spin system from equilibrium some-
how (preparation), to allow it to evolve after the
perturbation for various periods (evolution), and then to
detect what has happened in the evolution period by col-
lecting the subsequent FID (detection). If the evolution
is spin relaxation, we can measure the spin relaxation
time. In practice, however, the FID decays more rapidly
than expected from the true spin relaxation time. The
main cause is the spatial inhomogeneity of the external
magnetic field. In this case, the spin echo technique al-
lows us to avoid the artifact. The idea of spin echoes
is embedded in various schemes of pulse NMR experi-
ments. Since there are too many variations of 1-D-NMR
having their own acronyms, only two schemes are pre-
sented here to touch on some of the ideas.
Spin Decoupling. If we can determine whether or not
a pair of neighboring signals are really coupled to each
other, this method allows the assignment of the signals.
Also if the doublet splitting can be suppressed, it al-
lows the simplification of complex spectra. Consider
two spins A and X that are J-coupled with a coupling
constant J. If the spin X is irradiated selectively but
strongly with a microwave pulse at the resonance fre-
quency, the resonance A loses its doublet splitting (spin
decoupled) because the spin X flips so rapidly that the
A spin feels only the average of the spin states of X.
If we examine the presence or the absence of such spin
decoupling effects for pairs of doublets in a complex
NMR spectrum, we can identify J-coupled pairs within
a distance of 3–4 bonds in the molecular network.
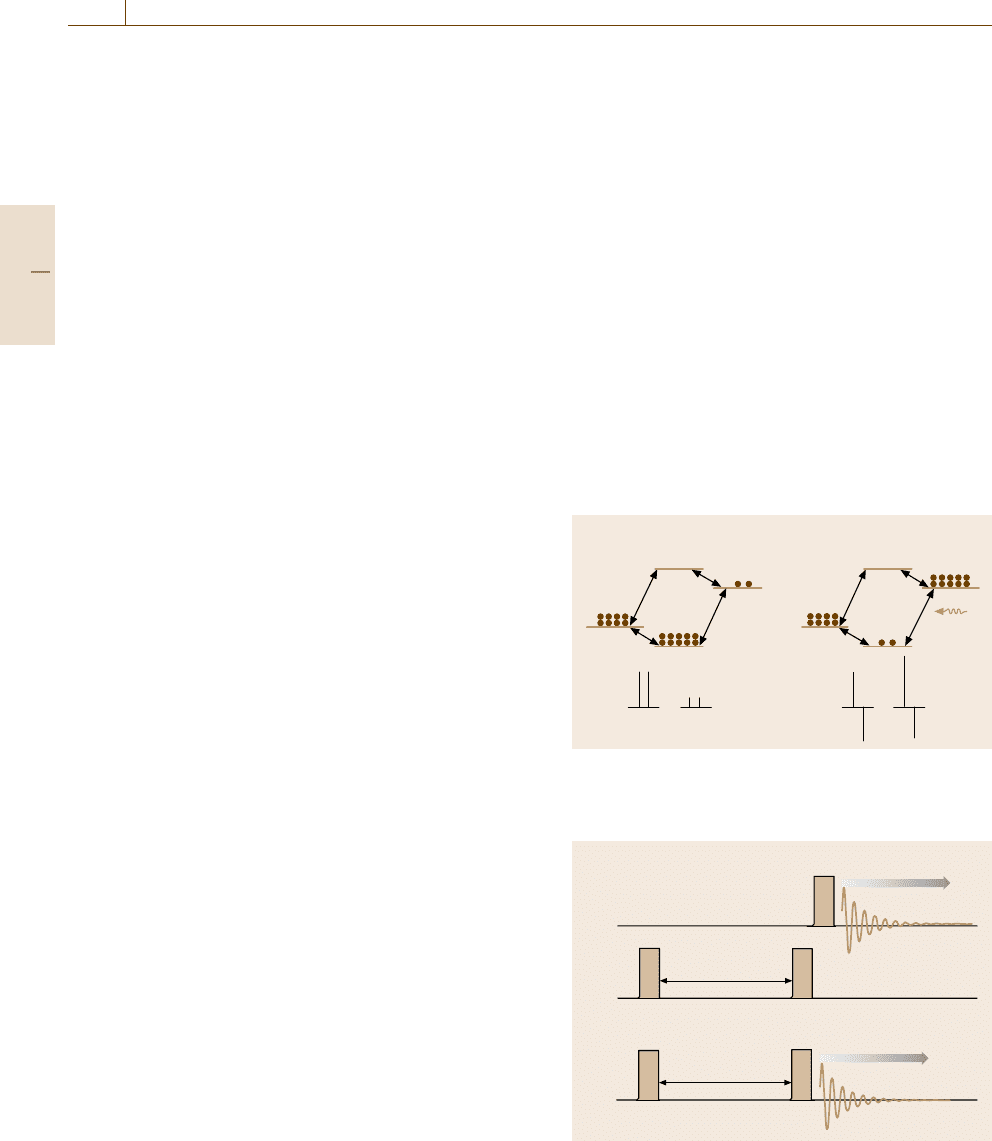
Selective Polarization Inversion (SPI). Another exam-
ple is given by again considering a J-coupled pair of
spins, A and X, but in this case, the pair is supposed
to be heteronuclear, e.g., A is
13
CandXis
1
H, as il-
lustrated by an energy level diagram in Fig. 5.88a. The
level notation βα, for example, indicates that the A
(
13
C) spin is down (β) while the X (
1
H) spin is up (α).
Due to the large difference in γ values between A and
X, the level population in thermal equilibrium differs
considerably. The bottom diagram in Fig. 5.88ashows
the schematic NMR spectrum with the vertical bars rep-
resenting the intensity. Now, consider that the resonance
between αα and αβ is selectively excited by a soft pulse
so as to invert the populations, as shown in Fig. 5.88b.
Then the NMR signals are drastically changed, as il-
lustrated by the bottom spectrum, because the signal
intensity is determined by the population difference.
The negative signal means that the FID component is
out of phase by π with the positive signal. Such changes
would not be observed if A and X are completely
independent, which signifies that the two spins are cou-
pled somehow. Unlike the NOE effect, the change is
immediate in the case of J-coupling, so we can dis-
tinguish whether they are dipole-coupled or J-coupled.
A subsidiary merit of polarization transfer techniques
is that weak NMR signals, such as that of
13
C, can be
enhanced.
Two-Dimensional (2-D) NMR. In small molecules, sig-
nal assignment may be possible through 1-D-NMR
experiments, but in large molecules such as proteins
that yield bewilderingly complex spectra, we need
more sophisticated NMR measurement schemes. Two-
dimensional NMR (2-D-NMR) is a successful solution
to this problem. The idea is to sort out crowded NMR
signals in a two-dimensional map according to which
pairs of signals are coupled to each other. The methods
may be regarded as double resonance techniques that
can be extended to multidimensional NMR by using ap-
a) b)
13
CΠ
1
H
H
2
C
2
H
1
C
1
H
2
C
2
H
1
H
2
H
1
C
1
C
2
C
1
H
2
H
1
C
2
C
1
ββ
αβ
βα
ββ
βα
αα
αβ
αα
Fig. 5.88a,b Heteronuclear selective polarization inver-
sion (SPI). (a) In thermal equilibrium, (b) when
1
His
selectively excited
a)
b)
t
2
AQ
π/2 pulse
π/2 pulse
π/2 pulse
t
1
π/2 pulse
1
H
t
1
t
2
π/2 pulse
1
H
AQ
13
C
Fig. 5.89a,b Pulse sequences for 2-D-NMR; (a) heteronu-
clear 2-D-NMR,
(b) homonuclear 2-D-NMR (COSY)
Part B 5.4
Nanoscopic Architecture and Microstructure 5.4 Molecular Architecture Analysis 265
propriate relays of pulse sequences. Two representative
schemes that are most commonly used are explained
briefly.
An example of pulse sequence diagram for 2-D-
NMR experiments is shown in Fig. 5.89a, in which two
heteronuclear spins of
13
Cand
1
H are assumed to be
J-coupled. Before FID is detected for
13
C spins, two
π/2 pulses separated by a time t
1
are applied to the
1
H
spins. Figure 5.90 depicts what happens in the vector
model. After the magnetization of
1
H is turned to the
y
-axis by the first π/2 pulse, the
1
H spins start to ro-
tate in the rotating frame at a rate Ω
H
= δ
H
± J
CH
/2,
where δ
H
is the chemical shift of the
1
H spins and J
CH
is the constant of J-coupling with
13
C, provided that
the reference frequency is set at the Larmor frequency
of the isolated
1
H. The diagrams in the second column
indicate this rotation at time t
1
increasing downward.
By the second π/2 pulse, the
1
H spins have turned fur-
ther around the x
-axis, as shown in the third column.
At this moment, the FID of
13
C spins, not of
1
H, is ac-
quired by applying a π/2 pulse on
13
C. When t
1
= 0,
the magnetization of
1
H is inverted, so the
13
C signal
changes drastically reflecting the inverted population
of
1
H, as in the case of SPI experiments in Fig.5.88 (the
x
-component, not turned by the second π/2 pulse, does
not affect the J-coupling). At the arbitrary time t
1
,the
magnetization of
1
H after the second π/2 pulse on
1
H
has a z-component of magnitude M cos Ω
H
t
1
,asshown
in the second line of the third column in Fig. 5.90,
so the
13
C signal intensity (schematically shown in
the right column) is modulated accordingly. Since the
13
C signal modulation has a period of 2πΩ
−1
H
,ifwe
repeat such measurements systematically changing the
time t
1
, the Fourier transform of the
13
C signal inten-
sity with respect to t
1
gives the NMR spectrum of the
1
H spins with a chemical shift δ
H
and the doublet split-
ting magnitude J
CH
. This may be regarded as a double
resonance or
13
C-NMR-detected NMR of
1
H spins. In
other words, the
13
C resonance signal acts as though
it is labeled with the resonances of the
1
H spins that
are J-coupled to the
13
C. If the π/2 pulses on
1
H are
hard and hence nonselective, we will obtain an NMR
spectrum in two dimensions for frequency ν
1
(corre-
sponding to t
1
) and for frequency ν
2
(corresponding to
t
2
), as illustrated in Fig. 5.91. The signals on the diag-
onal line (diagonal peaks) are always present, but the
off-diagonal signals (cross peaks) indicate the presence
of J-coupling between the corresponding
13
C nuclei
and
1
H nuclei. It is obvious that, even if the 1-D spectra
are complicated, the connectivity through J-coupling is
more easily resolved in such 2-D spectrum.
z
Mcos Ω
H
t
1
Mcos Ω
H
t
1
ν
2
Ω
H
= δ
H
±J
CH
/2
x'
y'
z
x'
y'
z
x'
y'
z
x'
y'
z
x'
y'
z
x'
y'
x'
y'
z
x'
y'
z
x'
y'
π
2
π
2
Fig. 5.90 Vector model for heteronuclear 2-D-NMR. For details,
see the text
The homonuclear version of the above scheme is
called correlated spectroscopy (COSY) and is widely
used to find J-couplings between
1
H nuclei. The pulse
sequence, shown in Fig. 5.89b, is similar to that in
Fig. 5.89a, except that the FID measured is now for
1
H
itself. Although the precise interpretation needs a quan-
tum mechanical description, the cause of the cross
peaks may be inferred by analogy to the heteronuclear
case above. Figure 5.92 shows an example of COSY
δ
H
δ
C
ξ
2
ξ
1
J
CH
J
CH
Cross peaks
Diagonal peaks
Fig. 5.91 Schematic 2-D-NMR spectrum of
13
Cand
1
H
J-coupled with each other
Part B 5.4
266 Part B Chemical and Microstructural Analysis
1
H (ppm)
1
H (ppm)
1.5
1.5
2.0
2.5
3.0
3.5
4.0
4.5
2.02.53.03.54.0
Fig. 5.92 DQF (double quantum-filtered)-COSY spectrum
of cyclic-GRGDSPA peptides (Courtesy of Prof. I. Shi-
mada)
(a more sophisticated double-quantum-filtered COSY)
spectrum obtained for the same peptide molecules
whose 1-D spectrum was shown in Fig. 5.85.
In the 2-D-NMR experiments discussed so far, the
pulse sequence generally consists of preparation, evo-
lution and detection stages. In Fig. 5.89a, the first π/2
pulse corresponds to preparation, the duration t
1
until
the second π/2 pulse to evolution, and the acquisi-
tion of FID to detection. In more sophisticated schemes
z
a)
e)
b)
f)
c)
g)
d)
h)
M
I
cos Ω
I
t
1
M
S
cos Ω
I
t
1
t
1
M
I
t
m
M
S
M
I
M
S
M
I
M
S
M
I
M
S
M
I
M
S
M
I
M
S
M
S
M
I
x'
y'
z
x'
y'
z
x'
y'
z
x'
y'
z
x'
y'
z
x'
y'
z
x'
y'
z
x'
y'
π
2
t
2
π
2
π
2
Fig. 5.94a–h Vector model of NOESY
Preparation
Evolution
Mixing
Detection
π/2 pulse π/2 pulseπ/2 pulse
t
1
t
m
t
2
1
H
AQ
Fig. 5.93 The pulse sequence for NOESY experiments
of 2-D-NMR, we add another stage, mixing, follow-
ing evolution. Figure 5.93 shows the pulse sequence
in NOESY (NOE spectroscopy) extensively used for
analysis of macromolecules. The corresponding vec-
tor model is shown in Fig. 5.94. The sequence of the
first π/2 pulse (Fig. 5.94a,b), an evolution time t
1
(Fig. 5.94b,c), and the second π/2 pulse (Fig. 5.94c,d)
is the same as in Fig. 5.90 except that we consider two
spins, I and S, having different resonance frequencies
but coupled through a dipole–dipole interaction. During
the mixing stage, which lasts for t
m
(Fig. 5.94e,f), the
z-components of the magnetization (the x
-components
have no effect and are not shown), which arise from
the population difference of the spin states, exchange
their populations due to the NOE effect. Thus, the inten-
sity of the NMR signals measured in the detection stage
(Fig. 5.94f,g) tells us how rapidly the relaxation has oc-
curred. Similarly to the COSY spectrum, the NOESY
spectrum is displayed with one axis representing the fre-
quencies of the I spin and another axis the frequencies
of the S spin. The cross peaks indicate that the two nu-
clei giving the signals are closer than ≈ 5 Å in space so
that they are dipole–dipole coupled.
Part B 5.4
Nanoscopic Architecture and Microstructure 5.4 Molecular Architecture Analysis 267
Sample Requirements. Finally, some comments are
added on the requirements for samples. Since NMR
signals are proportional to the number of spins, for
a sufficiently high signal-to-noise ratio to be achieved,
the sample amount must usually be greater than sev-
eral milligrams or 1 mM, which may be difficult. As
mentioned before, nuclei with spin I > 1/2 are not
very suitable for NMR experiments because the spec-
trum broadens due to the strong quadrupole interaction
(Sect. 5.1.2). Even if we confine ourselves to I = 1/2
nuclei, however, there are other causes of spectral
broadening that prohibit us from conducting high-
resolution NMR measurements. In actual experiments,
an inhomogeneity in the external magnetic field induces
an apparently rapid decay in the FID or a broadening
of the NMR spectra. Although the effect of magnetic
field inhomogeneity could be removed by the spin echo
technique, there still remains a cause of spectral broad-
ening due to the variation of molecular orientations,
which gives rise to different degrees of anisotropic
shielding and as a result continuously varying chem-
ical shifts. When the molecules are so small that they
rotate rapidly in the medium, the variation of chemical
shifts is averaged and consequently the NMR spectral
signals become sharpened. While small molecules in
solution benefit from this motional narrowing effect,
large molecules cannot do so if their rotation rate is too
small. Thus, NMR analysis generally becomes difficult
for large molecules that lack mobility. Although many
techniques, such as sorting signals according to inten-
tionally controlled spin relaxation lifetime, have been
developed to escape the difficulty, an upper limit exists
around 3 × 10
4
in molecular weight for which struc-
tural analysis by NMR is practically possible. This is
in great contrast to x-ray diffraction methods in which
much larger molecules, such as ribosome (4 × 10
6
in
molecular weight), can in principle be analyzed if single
crystals can be grown.
5.4.3 Chemophysical Analysis
Chromatography
Chromatography is a generic term for techniques
that separate complex mixtures into their components,
which are distributed with a variable probability be-
tween a stationary and a mobile phase; the methods are
based on the percolation of the mobile phase through
the solid phase in what is known as a column. The mo-
bile phase is gaseous in gas chromatography (GC)and
is liquid in liquid chromatography (LC). There are var-
ious schemes for chromatography further depending on
the type of stationary phase (solid or liquid) and hence
on the principle of molecular separation (ion exchange,
affinity difference, gel-filtration, hydrophobic interac-
tion etc.).
As illustrated in Fig. 5.95, chromatographic meas-
urements are conducted by constantly injecting into
a column a sample mixture carried by a mobile phase
and recording a chromatogram, a chart that plots the
amount of an analyte reaching a detector placed at the
outlet of the column versus the time t starting from the
sample injection. The retention time, t
r
, is the time be-
tween sample injection and a peak in the chromatogram,
while t
m
is the time taken for the mobile phase just to
pass through the column. The molecular species can
be identified from their retention time and their to-
tal amount is measured from the integrated area of
the corresponding peak. Among the various types of
detectors, the most common are thermal conductive de-
tectors, which are sensitive to any species. Using a mass
spectrometer as the detector, one can obtain more struc-
tural information of the separated molecular species.
GC is suitable for routine analysis and has high reso-
lution, short measurement time, low cost, and requires
a small amount of sample (1–10 μl in a liquid sam-
ple and 0.2–10 ml in a gaseous sample) as long as the
boiling temperature of the sample is below 300
◦
C; LC
is applicable to multicomponent, less volatile or py-
rolitic (thermally decomposable) samples that are not
covered by GC. The limitation of ordinary chromatogra-
phy is that peaks in chromatograms have no significant
structure, unlike those observed in photospectroscopy
methods that provide detailed information on the possi-
ble variety of each components. Nevertheless, there is
room for further improvements in detection techniques
Time
Analyte A Detector
Mobile phase
Stationary phase
A
mobile
A
stationary
Detected
signal
t
r
t
m
Fig. 5.95 General experimental configuration of chro-
matography
Part B 5.4