Czichos H., Saito T., Smith L.E. (Eds.) Handbook of Metrology and Testing
Подождите немного. Документ загружается.

1078 Part E Modeling and Simulation Methods
25
20
15
10
5
0
35
30
25
20
15
10
5
0
–20 –15 –10 –5 0 5
Total density of states
Total density of states
Energy E (eV)
–20 –15 –10 –5 0 5
Energy E (eV)
E
F
E
F
Ni
3
Al (DO
3
)
Co
3
Al (DO
3
)
A(Al)
2000
1800
1600
1400
1200
1000
800
600
400
Ni 20 40 60 80 Al
Temperature T(K)
Al (mol. %)
L
A(Ni)
A
NiAl
3
Ni
2
Al
3
Ni
5
Al
3
DO
3
+B2
Fink
Alexander
Phillips
Taylor
Nash
Robertson
Hilpert
Verhoeven
Jia
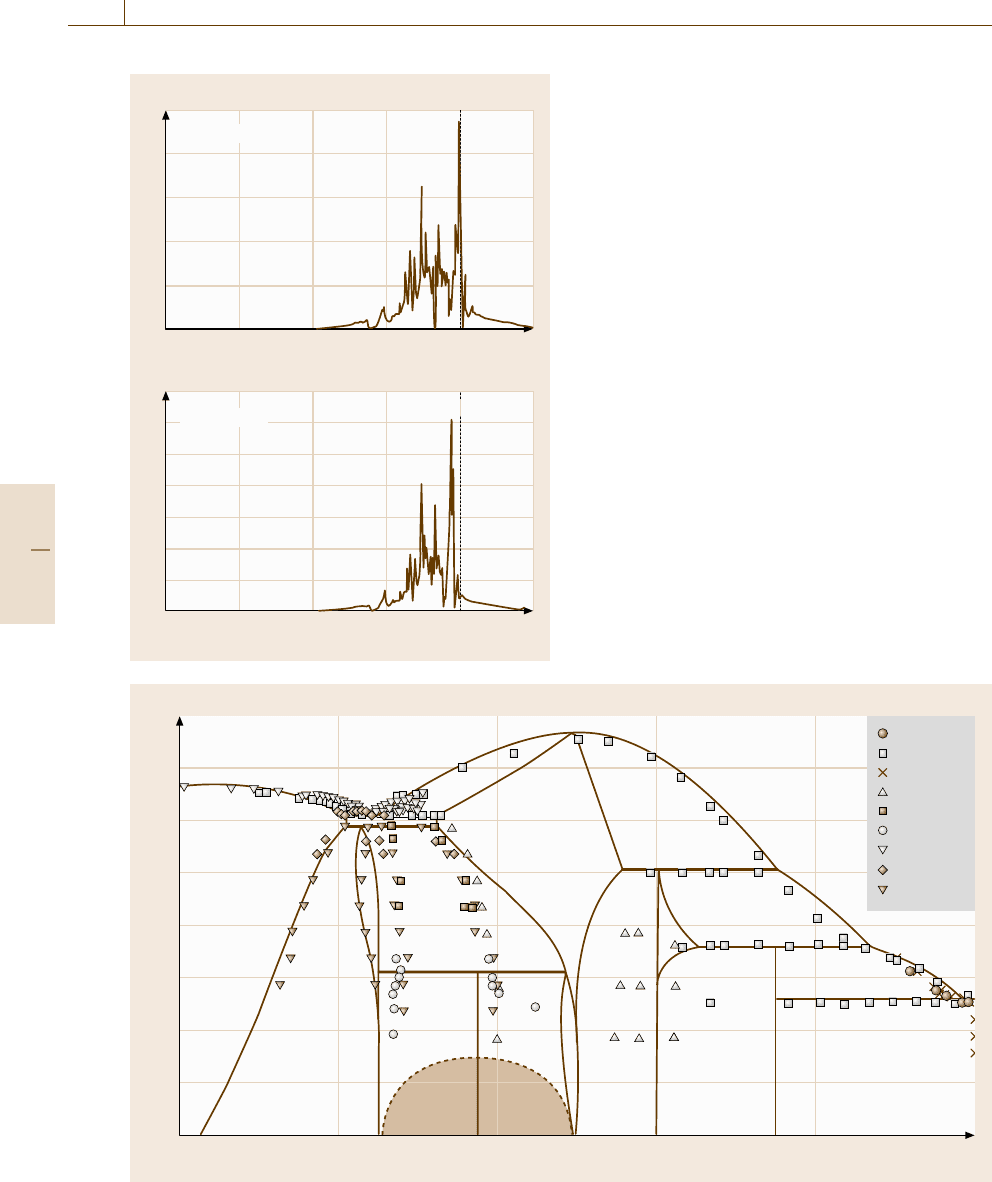
Fig. 20.17 The density of states of the D0
3
structure in the
Co
−
Al and Ni
−
Al alloy systems
This difference in the density of states at the Fermi
level might be considered to be the result of the ex-
tra d-electron in Ni versus Co. From the point of view
of the rigid band approximation, this difference in the
number of electrons for two neighboring elements shifts
the Fermi level towards the higher energy side in the
Ni
−
Al system versus the Co
−
Al system. This raises
the different relative positions of the Fermi level in the
DOS curve, which consequently leads to the different
structural stabilities.
The ground state analysis of the Ni
−
Al system
suggests that the phase separation concerning the D0
3
structure forms at absolute zero. When assuming that
such a two-phase separation forms at a finite tempera-
ture, however, the agreement between the calculated
phase boundaries and experimental data is insufficient.
For example, Fig.20.18 shows the phase diagram in the
case that a two-phase separation between the metastable
D0
3
and B2 structures occurs around 500
◦
C. The
homogeneity range of the B2 single phase is compar-
Fig. 20.18 The calculated Ni
−
Al phase diagram assuming
that the critical temperature of the two-phase separation be-
tween the metastable D0
3
and B2 structures was around
500
◦
C
Part E 20.2

The CALPHAD Method 20.3 Prediction of Thermodynamic Properties of Compound Phases with First-principles Calculations 1079
atively reduced owing to the influence of the metastable
miscibility gap with decreasing temperature, and ac-
cordingly, the calculated phase boundaries deviate from
the experimental data. Therefore, the phase separation
of the D0
3
structure is not likely in the experimentally
observable temperature range.
20.3 Prediction of Thermodynamic Properties of Compound Phases
with First-principles Calculations
In an analysis of phase equilibria containing compound
phases, physical properties of metastable structures are
often required. The necessity appears clearly for in-
stance in the following case. Figure 20.19 shows the
isothermal phase diagram for the Fe
−
Cr
−
C ternary
system. In this ternary system, several types of carbides
form in which some amount of alloying element is sol-
uble. If we consider the cementite, Cr substitutes more
than 10% of Fe, and it forms the ternary line compound.
In such a case, the Gibbs energy of the cementite phase
is usually described by using sublattice model as the
(Fe, Cr)
3
C formula. Then if we want to evaluate the
thermodynamic function for this phase, we need the
formation energy of Cr
3
C, which is metastable in the
Cr
−
C binary system. In the procedure of CALPHAD
approach, this parameter is usually determined on the
basis of the experimental data in the Fe-rich side. How-
ever, it could be easily understood that this technique
follows large amount of errors in estimation. Applying
the first-principles calculation may possibly solve this
difficulty. Thus in the present section, some examples
for application to predicting thermodynamic properties
of compound phases and phase diagram calculations
will be illustrated.
Carbides and nitrides play a key role in the micro-
structure control of steels, due to a fine disper-
sion of these precipitates. The effectiveness of the
first-principles calculations to the analysis of thermo-
dynamic properties of these compounds might be an
interesting issue. A comparison of the calculated forma-
tion energies with the experimental values is attempted
for some typical carbides observed in steels to clarify
the validity of the FLAPW method. Table 20.8 [20.18]
shows the formation energy, ΔE
φ
form
,definedbyaver-
aging the total energy of the constituent elements with
chemical composition up to the segregation limit, as
follows
ΔE
φ
form
= E
φ
tot
−x
φ
M
E
M
tot
−
1 −x
φ
M
E
C
tot
, (20.37)
where φ denotes the type of carbide, and M and C rep-
resent a metallic element and graphite, respectively. For
example, the formation energy of Fe
3
C in the paramag-
netic state is calculated to be 17.9kJ/mol, while the
formation energy of Fe
3
C, by considering the spin po-
larization, is 8.1kJ/mol. This result shows the effect
of the ferromagnetism of the Fe
3
C phase in the lower
temperature region. Furthermore, because the formation
energy from bcc-Fe and graphite is positive, the Fe
3
C
structure is less stable than graphite at absolute zero.
The calculated formation energies for the Cr
7
C
3
and
Cr
23
C
6
phases show a reasonable agreement with the
thermodynamic data reported in the literature [20.19].
From consideration of the data shown in Table 20.8,the
thermodynamic properties for metallic carbides evalu-
ated by the first-principles calculations can be applied to
the general procedures used in the CALPHAD method.
20.3.1 Thermodynamic Analysis
of the Fe–Al–C System
The Perovskite carbide in this ternary system, Fe
3
AlC
(κ), is an fcc-based ordered phase with an E2
1
-type
90
80
70
60
50
40
30
20
10
C
Fe Cr
Cr (at. %)
C (at. %)
20 40 60
α +M
23
C
6
γ+ M
7
C
3
M
7
C
3
+C
80
γ
Fe
3
C
M
7
C
3
Cr
3
C?
M
23
C
6
Fig. 20.19 The isothermal phase diagram for the Fe
−
Cr
−
Cternary
system
Part E 20.3
1080 Part E Modeling and Simulation Methods
Table 20.8 Comparison of the calculated formation energies with the experimental values for some typical steel carbides
Carbides Space Calculated lattice Observed lattice Calculated formation Observed formation
group parameter (nm) parameter (nm) enthalpy in the ground enthalpy at 25
◦
C
state (kJ/mol) (kJ/mol)
Fe
3
C Pnma a =0.4871 − +17.9 −
(paramagnetic) b =0.6455 − −
c =0.4330 − −
Fe
3
C Pnma a =0.5018 a =0.5078 +8.1 +6.3
(ferromagnetic) b =0.6650 b =0.67297
c =0.4460 c =0.45144
Cr
7
C
3
Pnma a = 0.4373 a =0.4526 −19.8 −22.8
b =0.6772 b =0.7010
c =1.1730 c =1.2142
Cr
23
C
6
Fm-3m a =1.0475 a = 1.06595 −21.8 −19.7
structure. The application of this material as a heat
resistant alloy from the formation of a coherent fine
microstructure consisting of an fcc solid solution and
the κ phase is attracting great attention [20.20]. Regard-
less of such a promising potential for this new material,
little information on the thermodynamic properties of
this carbide phase is known. Thus, an attempt to calcu-
late the full phase equilibria of the Fe
−
Al
−
C ternary
system is made, introducing the first-principles val-
ues for Fe
3
AlC into a CALPHAD-type thermodynamic
analysis.
In the Fe
3
AlC structure, the Fe and Al atoms are ar-
rangedinanL1
2
-type superlattice, in which the C atoms
occupy interstitial sites, resulting in an E2
1
superstruc-
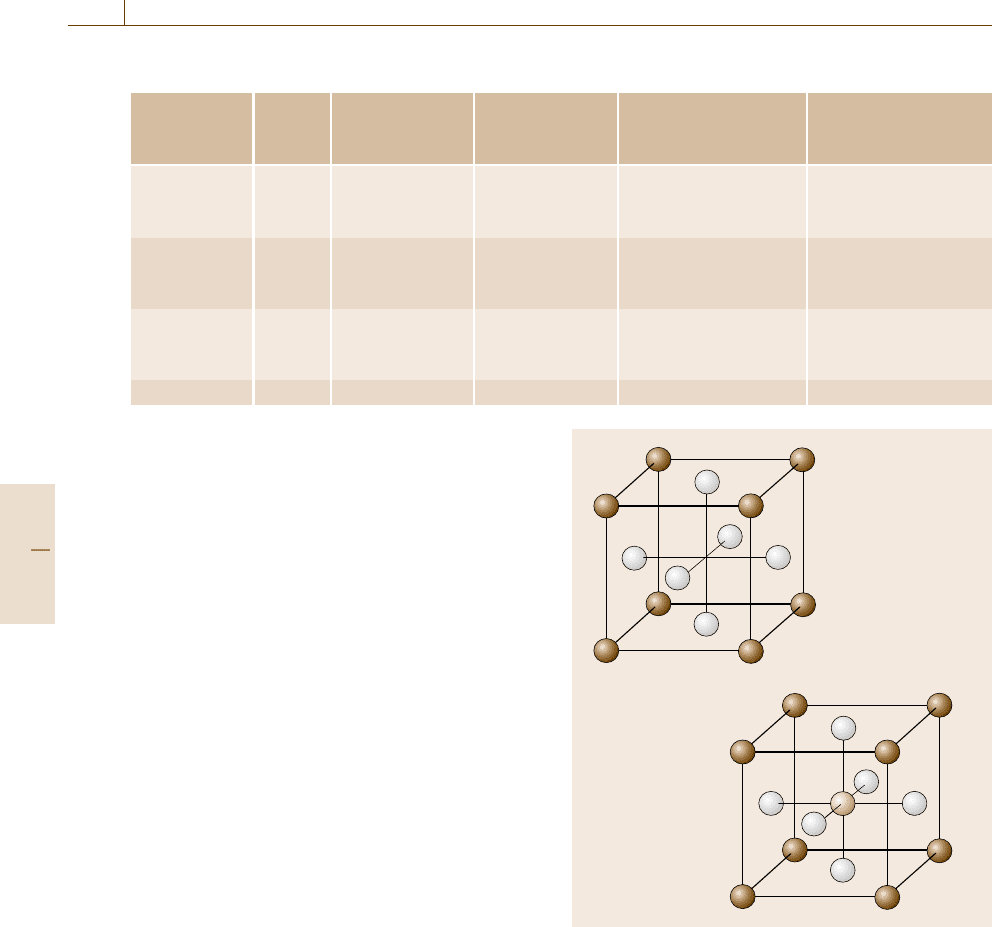
ture. Figures 20.20a, 20.20b show schematic diagrams
of the Fe
3
Al–L1
2
and Fe
3
AlC–E2
1
structures, respec-
tively. If the C atom occupies only the body-centered
sites, then it is surrounded by six Fe atoms, and this is
preferable from an energetic point of view. Occupation
of the other fcc interstitial sites enhances the tetrag-
onality of the L1
2
crystal structure, yielding a larger
strain energy. The only difference between these two
structures is the existence of C atoms in the octahedral
interstitial sites. Therefore, the Gibbs free energies of
these two ordered structures should be described by the
same thermodynamic model. The two-sublattice model
denoted by the formula,
Fe
y
(1)
Fe
Al
y
(1)
Al
3
Fe
y
(2)
Fe
Al
y
(2)
Al
1
,
was generally applied to the L1
2
structure, and the
three-sublattice model,
Fe
y
(1)
Fe
Al
y
(1)
Al
3
Fe
y
(2)
Fe
Al
y
(2)
Al
1
C
y
(3)
C
Va
y
(3)
Va
1
,
Fe
3
Al-L1
2
Al
Fe
Fe
Al
C
Fe
3
a)
b)
AlC-E2
1
Fig. 20.20a,b Crystal structures for (a) Fe
3
Al–L1
2
and
(b) Fe
3
AlC–E2
1
is applied to the κ phase. The Gibbs free energy for the
κ phase is calculated using (20.38):
G
κ
= y
(1)
Al
y
(2)
Al
y
(3)
C
0
G
κ
Al:Al:C
+y
(1)
Al
y
(2)
Al
y
(3)
Va
0
G
κ
Al:Al:Va
+y
(1)
Al
y
(2)
Fe
y
(3)
C
0
G
κ
Al:Fe:C
+y
(1)
Al
y
(2)
Fe
y
(3)
Va
0
G
κ
Al:Fe:Va
+y
(1)
Fe
y
(2)
Al
y
(3)
C
0
G
κ
Fe:Al:C
+y
(1)
Fe
y
(2)
Al
y
(3)
Va
G
κ
Fe:Al;Va
Part E 20.3

The CALPHAD Method 20.3 Prediction of Thermodynamic Properties of Compound Phases with First-principles Calculations 1081
+y
(1)
Fe
y
(2)
Fe
y
(3)
C
0
G
κ
Fe:Fe:C
+y
(1)
Fe
y
(2)
Fe
y
(3)
Va
0
G
κ
Fe:Fe:Va
+3RT
y
(1)
Al
ln y
(1)
Al
+y
(1)
Fe
ln y
(1)
Fe
+RT
y
(2)
Al
ln y
(2)
Al
+y
(2)
Fe
ln y
(2)
Fe
+RT
y
(3)
C
ln y
(3)
C
+y
(3)
Va
ln y
(3)
Va
+y
(1)
Al
y
(1)
Fe
y
(2)
Al
y
(3)
C
L
κ
Al,Fe:Al:C
+y
(1)
Al
y
(1)
Fe
y
(2)
Al
y
(3)
Va
L
κ
Al,Fe:Fe:Va
+y
(1)
Al
y
(1)
Fe
y
(2)
Fe
y
(3)
C
L
κ
Al,Fe:Fe:C
+y
(1)
Al
y
(1)
Fe
y
(2)
Fe
y
(3)
Va
L
κ
Al,Fe:Fe:Va
+y
(1)
Al
y
(2)
Al
y
(2)
Fe
y
(3)
C
L
κ
Al:Al,Fe:C
+y
(1)
Al
y
(2)
Al
y
(2)
Fe
y
(3)
Va
L
κ
Al:Al,Fe:Va
+y
(1)
Fe
y
(2)
Al
y
(2)
Fe
y
(3)
C
L
κ
Fe:Al,Fe:C
+y
(1)
Fe
y
(2)
Al
y
(2)
Fe
y
(3)
C
L
κ
Fe:Al,Fe:Va
+y
(1)
Al
y
(2)
Al
y
(3)
C
y
(3)
Va
L
κ
Al:Al:C,Va
+y
(1)
Al
y
(2)
Fe
y
(3)
C
y
(3)
Va
L
κ
Al:Fe:C,Va
+y
(1)
Fe
y
(2)
Al
y
(3)
C
y
(3)
Va
L
κ
Fe:Al:C,Va
+y
(1)
Fe
y
(2)
Fe
y
(3)
C
y
(3)
Va
L
κ
Fe:Fe:C,Va
. (20.38)
Table 20.9 Calculated thermodynamic parameters required by the (Fe, Al)
3
(Fe, Al)
1
(C, Va)
1
-type three-sublattice
model
Structure Structure Magnetism Calculated lattice Observed lattice Calculated formation
symbol parameter (nm) parameter (nm) enthalpy in the ground state
(kJ/mol of compound)
Fe
3
AlC E2
1
Paramagnetic 0.3677 − −128.5
Ferromagnetic 0.3677 0.3781 −139.5
Al
3
FeC E2
1
Paramagnetic 0.3890 − +190.5
Fe
3
Al L1
2
Paramagnetic 0.3502 − −35.2
FeAl
3
L1
2
Paramagnetic 0.3734 − −67.6
Fe
4
C − Paramagnetic 0.3645 − +88.4
Al
4
C − Paramagnetic 0.4057 − +160.5
Calculation of the Thermodynamic Properties
of the κ Phase
The thermodynamic parameters required by the model
in the (Fe, Al)
3
(Fe, Al)
1
(C, Va)
1
form are evaluated us-
ing first-principles calculations and the results are listed
in Table 20.9. The calculated values denote the forma-
tion enthalpies based on the stable structure of the pure
element in the ground state. The ferromagnetic state of
the κ phase is calculated to be almost 10 kJ/mol more
stable than its paramagnetic state, and the magnetic mo-
ment of the phase was calculated to be 3.05 μB. Besides
these cohesive energies for the stoichiometric com-
ponents, the interactions between atoms on the same
sublattice are defined in the same way as for the thermo-
dynamic parameters, and these interaction parameters,
as well as the entropy term of the formation energy, are
estimated using the experimental phase boundaries.
Electronic Structure and Phase Stability
of the κ Phase
As shown in Fig. 20.20, a crystallographic similarity
exists between the Perovskite carbide κ and the Fe
3
Al–
L1
2
structure, i. e., a C atom is placed in the center of
an octahedron composed of six Fe atoms occupying the
face-centered positions in the L1
2
structure. The actual
calculated equilibrium lattice constant of the Fe
3
Al–
L1
2
structure is 0.3502 nm, while that of the κ phase is
0.3677 nm, which correspond well with the experimen-
tal results of Palm and Inden [20.21]. This fact implies
that occupation by the C atoms in the octahedral inter-
stitial sites causes an expansion of the L1
2
lattice. Since
the calculated enthalpy of formation of the κ phase
( −27.9kJ/mol of atoms) is much lower than that of the
Fe
3
Al–L1
2
structure (−8.8kJ/mol of atoms), it is con-
cluded that the interstitial C atoms enhance the stability
of the L1
2
structure. Thus, the role of the C atoms will
be discussed in the context of their electronic structure.
Part E 20.3
1082 Part E Modeling and Simulation Methods
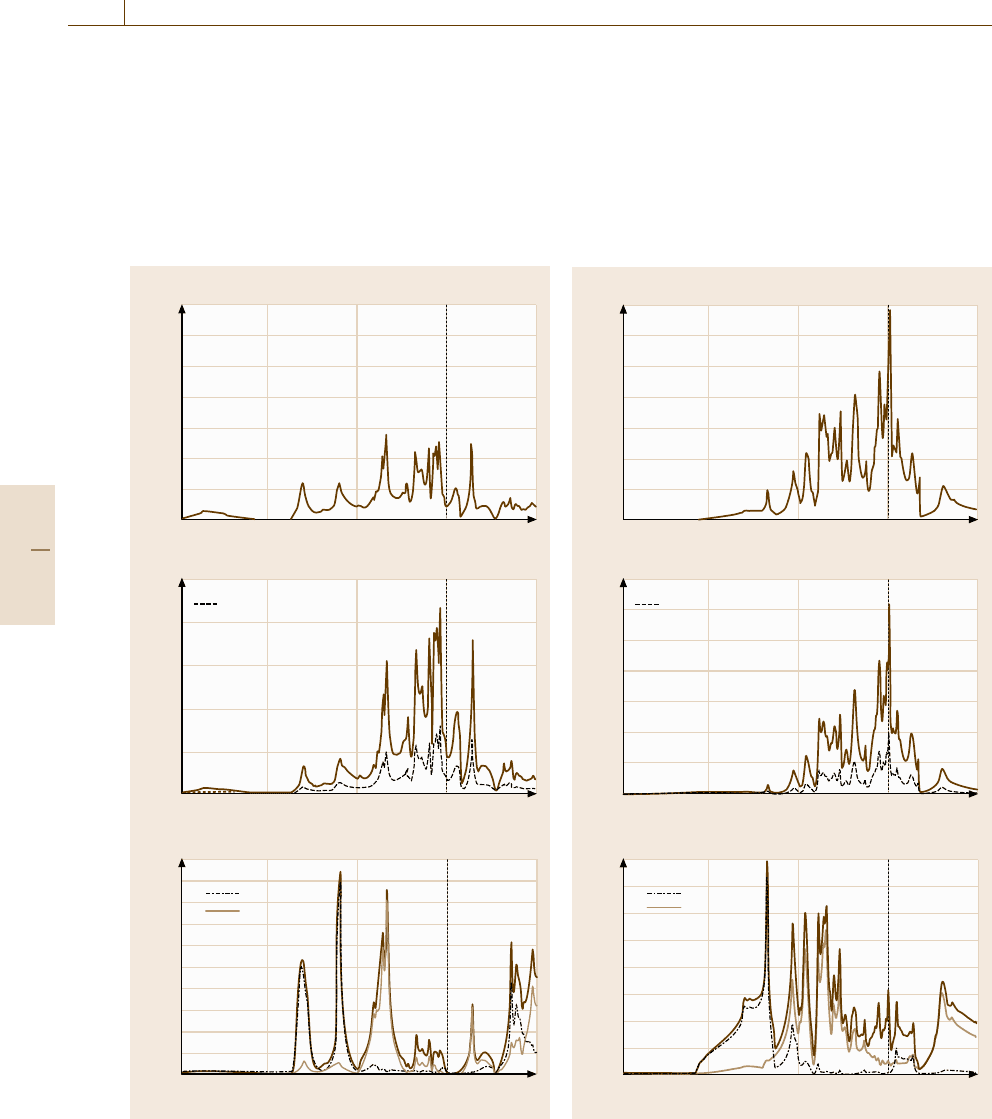
Figures 20.21 and 20.22 show the total density of
states (total DOS) and the angular-momentum-resolved
density of state for each element (p-DOS)forthe
Fe
3
AlC–E2
1
(κ)andFe
3
Al–L1
2
structures, respec-
tively. The term E
F
denotes the Fermi energy, and no
electrons occupy electronic states above this energy
level. In both structures, the DOS mainly consist of the
contribution from the Fe d-electrons. However, while
14
12
10
8
6
4
2
0
Fe
3
AlC-E2
1
4
3
2
1
0
0.16
0.12
0.08
0.04
0
–15 –10 –5
–15 –10 –5
–15 –10 –5 0 5
Fe
3
AlC-E2
1
Fe
3
AlC-E2
1
a) Total DOS
b) p-DOS (Fe)
c) p-DOS (Al)
(eV)
05
(eV)
05
(eV)
d-xy
s
p
E
F
Fig. 20.21 (a) Total density of states, and angular-
momentum-resolved density of states for: (b) Fe and (c) Al
for the Fe
3
AlC–E2
1
(κ) structure
observing these diagrams, we noticed a marked differ-
ence between these two structures, in that the Fermi
level is located near the peak of the total DOS for the
Fe
3
Al–L1
2
structure, but decreases in a region with
a very low DOS in the Fe
3
AlC–E2
1
structure. This fact
indicates that from an energetic point of view, the sta-
ble E2
1
structure of the κ phase is highly preferred,
compared with the Fe
3
Al–L1
2
structure.
14
12
10
8
6
4
2
0
Fe
3
Al-L1
2
12
10
8
6
4
2
0
0.3
0.2
0.1
0
–15 –10 –5
–15 –10 –5
–15 –10 –5 0 5
Fe
3
AlC-E2
1
Fe
3
AlC-E2
1
a) Total DOS
b) p-DOS (Fe)
c) p-DOS (Al)
(eV)
05
(eV)
05
(eV)
d-xy
s
p
E
F
Fig. 20.22 (a) Total density of states, and angular-
momentum-resolved density of states for: (b) Fe and (c) Al
for the Fe
3
Al–L1
2
structure
Part E 20.3
The CALPHAD Method 20.3 Prediction of Thermodynamic Properties of Compound Phases with First-principles Calculations 1083
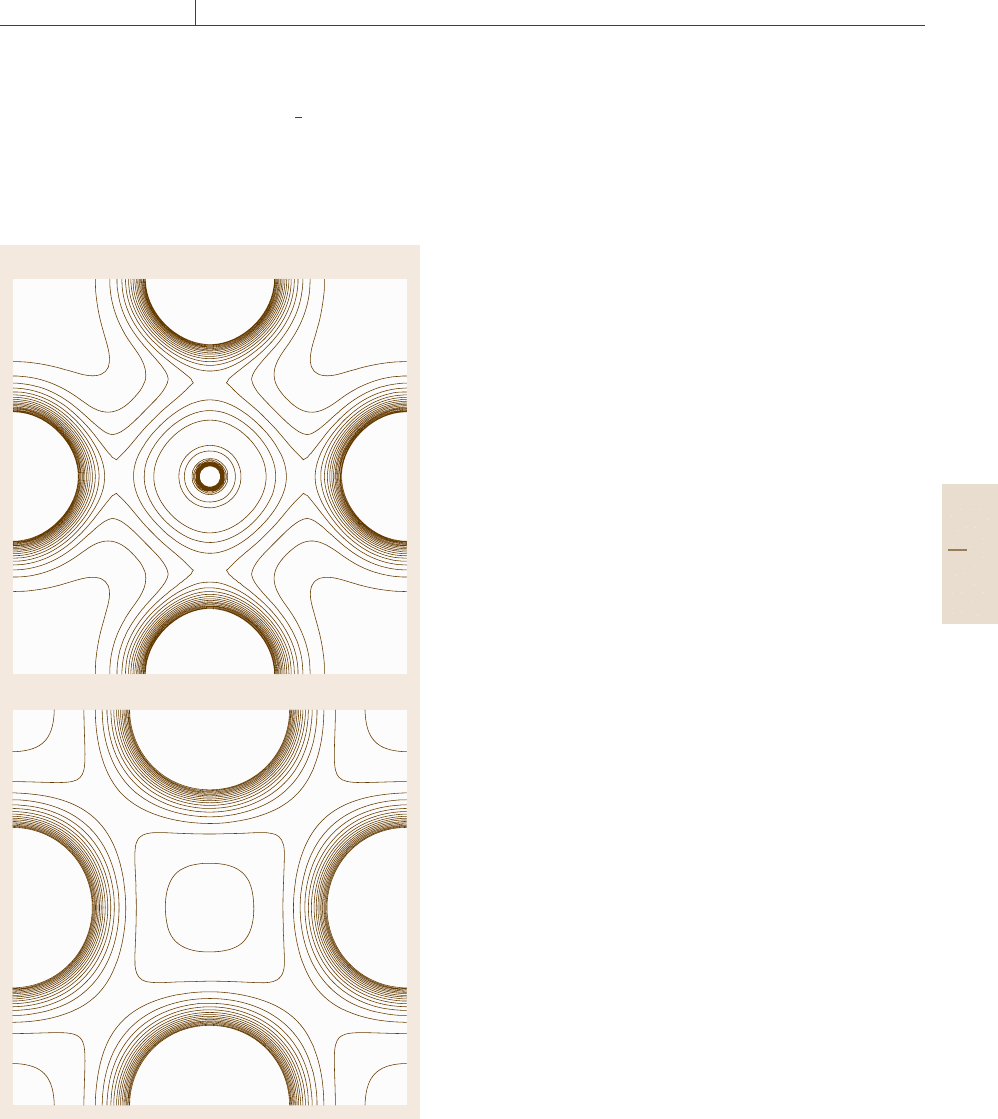
Figures 20.23aand20.23b show the calculated
electron charge density plots of the Fe
3
AlC–E2
1
(κ)
and Fe
3
Al–L1
2
structures in the (00
1
2
) plane, where
the contour lines correspond to an electron density of
100 e/nm
3
. The Fe atoms can be seen in the middle of
the horizontal and vertical axes, while the C atoms are
located in the center of the Fig. 20.23a. From these con-
a) Fe
3
AlC-E2
1
b) Fe
3
Al-L1
2
Fe
Fe
Fe
Fe
Fe
Fe
C
Fe
Fe
Fig. 20.23a,b Calculated electron charge density plots of:
(a) the Fe
3
AlC–E2
1
(κ)and(b) the Fe
3
Al–L1
2
structures
in the (001/2) plane
tour plots, it can be seen that bonding between the Fe
and C atoms occurs in the Fe
3
AlC–E2
1
structure, since
a finite charge density between these atoms can be ob-
served. This interaction between the atoms enhances the
energetic stability of the Fe
3
AlC–E2
1
structure.
Comparison of the Calculated Phase Equilibria
with the Experimental Data
The calculated Fe
−
Al
−
C ternary phase diagrams are
showninFig.20.24 for temperatures, T = 800, 1000,
and 1200
◦
C. The enlarged portion of the isothermal
section diagrams is compared with the experimental
phase boundaries determined using X-ray diffraction
and metallographic observation [20.21]. From EPMA
measurements, they reported that the chemical compo-
sition of the κ phase shifted remarkably from its stoi-
chiometric composition. The homogeneity range of the
κ phase extends from Fe
2.9
Al
1.1
C
0.7
to Fe
2.8
Al
1.2
C
0.7
according to the calculations.
20.3.2 Thermodynamic Analysis
of the Co–Al–C and Ni–Al–C Systems
The microstructures of Ni-based superalloys contain the
Ni
3
Al–L1
2
phase, which shows an anomalous flow–
stress dependence on temperature [20.22]. In addition,
because they exhibit very high melting temperatures
and have good resistance to oxidation, alloys with com-
plex phase structures containing the NiAl–B2, fcc-Ni,
and Ni
3
Al–L1
2
phases have been investigated for tech-
nological applications. On the other hand, it is difficult
to produce Co
−
Al-based superalloys with a microstruc-
ture consisting of fcc-Ni and Ni
3
Al–L1
2
, because of
the absence of a stable strengthening L1
2
phase in
the Co
−
Al binary system. However, the addition of
carbon to this alloy stabilizes the formation of the
Perovskite type carbide (E2
1
) with the composition,
M
3
AlC (κ phase), as seen in Fig. 20.18, and this carbide
is anticipated to form a fine coherent microstructure in
a Co-based solid solution. Then the entire phase equi-
libriaoftheCo
−
Al
−
C ternary system is attempted to
be clarified by coupling the CALPHAD and ab ini-
tio calculations. The same procedure is applied to the
Ni
−
Al
−
C system, and the results are compared to the
Co
−
Al
−
C system.
Calculation of the Co–Al–C Phase Diagram
The same thermodynamic model as (Fe, Al)
3
(Fe, Al)
1
(C, Va)
1
is applied to the Co
−
Al
−
CandNi
−
Al
−
C
based κ phase, and the parameters necessary for this
model are evaluated using first-principles calculations,
Part E 20.3
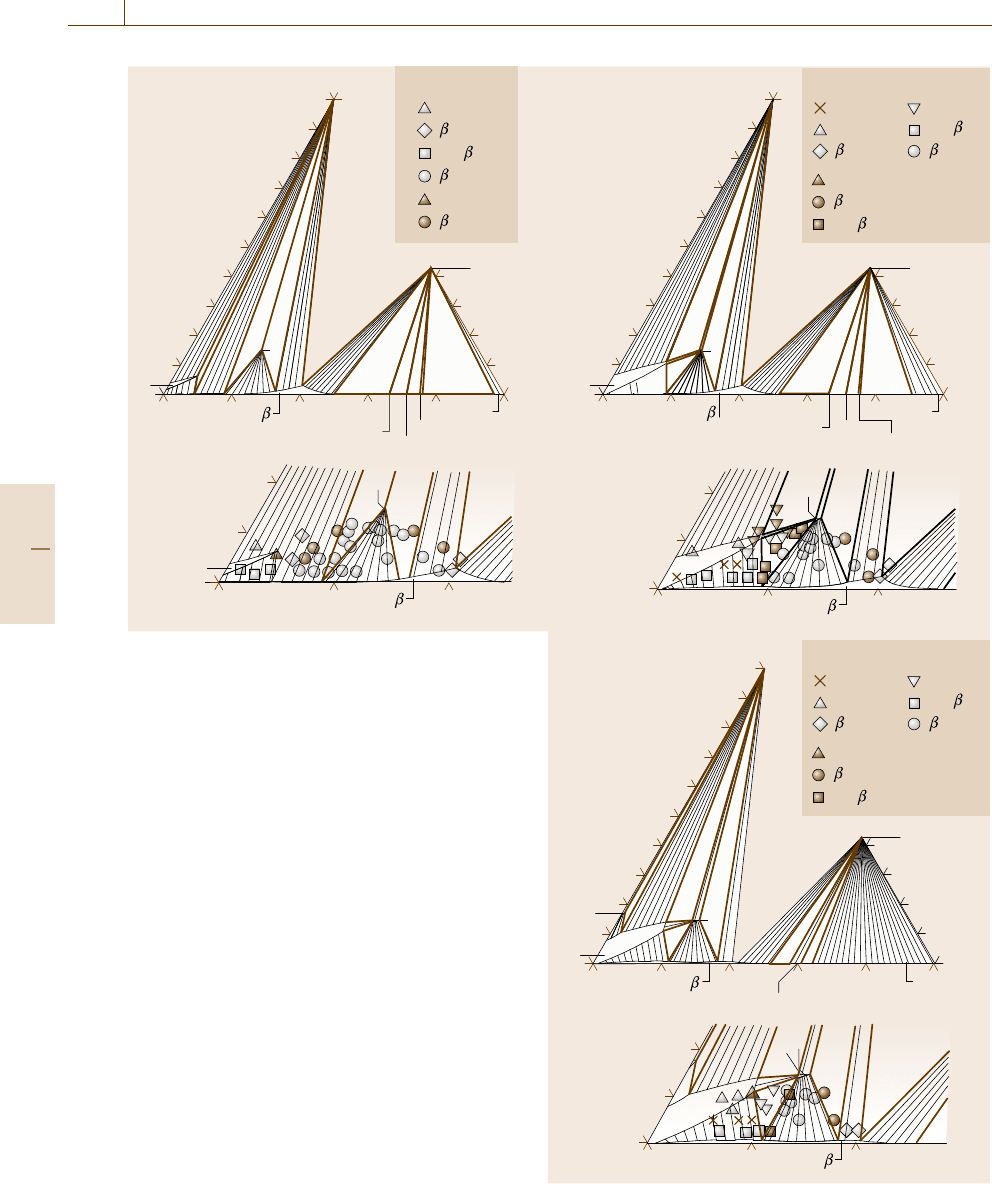
1084 Part E Modeling and Simulation Methods
Al
10
20
30
40
50
60
70
80
90
a) 800 °C
C
10
20
30
40
50
60
70
80
90
20 40 60 80Fe Al
Al
4
C
3
Fe
4
Al
13
FeAl
2
Fe
25
L
C content
(mol. %)
γ
κ
Fe
10
20
20 40
mol. % C
mol. % Al
κ
γ
Palm, Inden
γ
γ
κ
γ
κ
κ
+
+
+
+
+ (C)
+ (C)
+ (C)
+ (C)
b) 1000 °C
C
10
20
30
40
50
60
70
80
90
20 40 60 80Fe Al
Al
4
C
3
Fe
4
Al
13
FeAl
2
Fe
2
Al
5
L
C content
(mol. %)
γ
κ
Fe
10
20
20 40
mol. % C
mol. % Al
κ
η
Al content (mol. %)
c) 1200 °C
C
20 40 60 80Fe Al
Al
4
C
3
L
C content
(mol. %)
γ
κ
Fe
10
20
20
40
mol. % C
mol. % Al
κ
Palm, Inden
η
Al content (mol. %)
L
Fe
4
Al
5
γ
+ (C)
γ + (C)
κ
+
γ
+
γ
+
κ
γ +
κ
+
κ
+ + (C)
γ
κ
+ + (C)
Palm, Inden
γ
+ (C)
γ + (C)
κ
+
γ
+
γ
+
κ
γ +
κ
+
κ
+ + (C)
γ
κ
+ + (C)
Fig. 20.24a–c Isothermal section diagram calculated
at:
(a) 800
◦
C, (b) 1000
◦
C, and (c) 1200
◦
C with an
enlarged portion on the Fe-rich side
Part E 20.3

The CALPHAD Method 20.3 Prediction of Thermodynamic Properties of Compound Phases with First-principles Calculations 1085
Table 20.10 The calculated thermodynamic parameters required by the (M, Al)
3
(M, Al)
1
(C, Va)
1
-type three sublattice
model
Structure Structure Calculated lattice Observed lattice Calculated formation
symbol parameter (nm) parameter (nm) enthalpy in the ground state
(kJ/mol of compound)
Co
3
AlC E2
1
0.3675 0.3700 −179.0
Al
3
CoC E2
1
0.3909 − +113.5
Co
3
Al L1
2
0.3515 − −79.6
CoAl
3
L1
2
0.3726 − −90.4
Co
4
C − 0.3621 − +98.0
Al
4
C − 0.4057 − +160.5
Ni
3
AlC E2
1
0.3713 −141.0
Al
3
NiC E2
1
0.3905 +64.5
Ni
3
Al L1
2
0.3504 0.3572 −169.5
NiAl
3
L1
2
0.3774 −91.6
Ni
4
C − 0.3645 +61.0
as listed in Table 20.10. The calculated Co
−
Al
−
C
ternary phase diagrams are shown in Fig. 20.25 for
temperatures of 900
◦
C, 1100
◦
C, and 1300
◦
C. The
enlarged portion of the isothermal section diagrams
at 1100
◦
C was compared with the experimental
data [20.21].
The κ phase only appears at the stoichiometric com-
position in our results, while a small homogeneity range
is exhibited by this phase in the experimental phase
diagrams. This aspect is closely related to the large,
positive formation energies in the metastable ordered
structures, such as Al
3
CoC and Al
4
C. As the formation
energy of Co
3
AlC shows an extremely large negative
value when compared to these structures, the κ phase
only forms at the stoichiometric position. The homo-
geneity can be expressed by introducing interaction
parameters between unlike atoms in the same sublattice.
However, this treatment is not applied in the present
analysis, because the experimental phase boundaries of
the κ phase are still uncertain.
The calculated vertical section diagrams at constant
10 mol % C and 30 mol % Al are shown in Fig. 20.26b.
The calculated values agree well with the experimen-
tal results, and hence, the new type of approach based
on the incorporation of the CALPHAD method into ab
initio calculations has proven to be applicable to phase
diagram calculations for higher-order systems.
Calculation of the Ni–Al–C Phase Diagram
In the Ni
−
Al
−
C system, on the other hand, it has been
experimentally verified that the κ phase does not appear
in the vicinity of the stoichiometric composition. Fig-
ures 20.27a–c show the calculated isothermal section
diagrams at 900, 1000, and 1300
◦
C, respectively.
Phase Separation of the κ Phase
in the Co–Ni–Al–C Quaternary System
According to the first-principles calculations, there was
a large difference in the phase stability between the
Co
3
Al–L1
2
and Ni
3
Al–L1
2
phases, compared to the
difference between the Ni
3
AlC–E2
1
and Co
3
AlC–E2
1
phases. In such an energetic situation, a two-phase sep-
aration should occur, depending on the difference in
the formation energies of the compounds, given by the
following expression
ΔG =
0
G
Ni
3
Al
+
0
G
Co
3
AlC
−
0
G
Co
3
Al
−
0
G
Ni
3
AlC
.
(20.39)
This miscibility gap originates in the energy differ-
ence between the terminal compounds, and as the
absolute value of ΔG increases, then the critical
temperature of the miscibility gap increases. Such
a phase separation is often observed in some com-
plex carbonitrides or alloy semiconductor systems.
Figure 20.28 shows the calculation of a miscibility gap
in the Co
3
AlC
−
Ni
3
AlC
−
Co
3
Al
−
Ni
3
Al pseudoquater-
nary system at T =1000
◦
C. In this model calculation,
only the formation enthalpies of the stoichiometric com-
pounds are used, designated by the four vertices of the
composition square. One can see that a two-phase sep-
aration forms in the direction of the Co
3
AlC and Ni
3
Al
diagonal. Therefore, a two-phase separation between
these terminal compounds will be involved in the phase
equilibria of the quaternary system.
Using the thermodynamic description of the four
ternary systems that comprise the Co
−
Ni
−
Al
−
C qua-
ternary system, a vertical section diagram is calculated
Part E 20.3
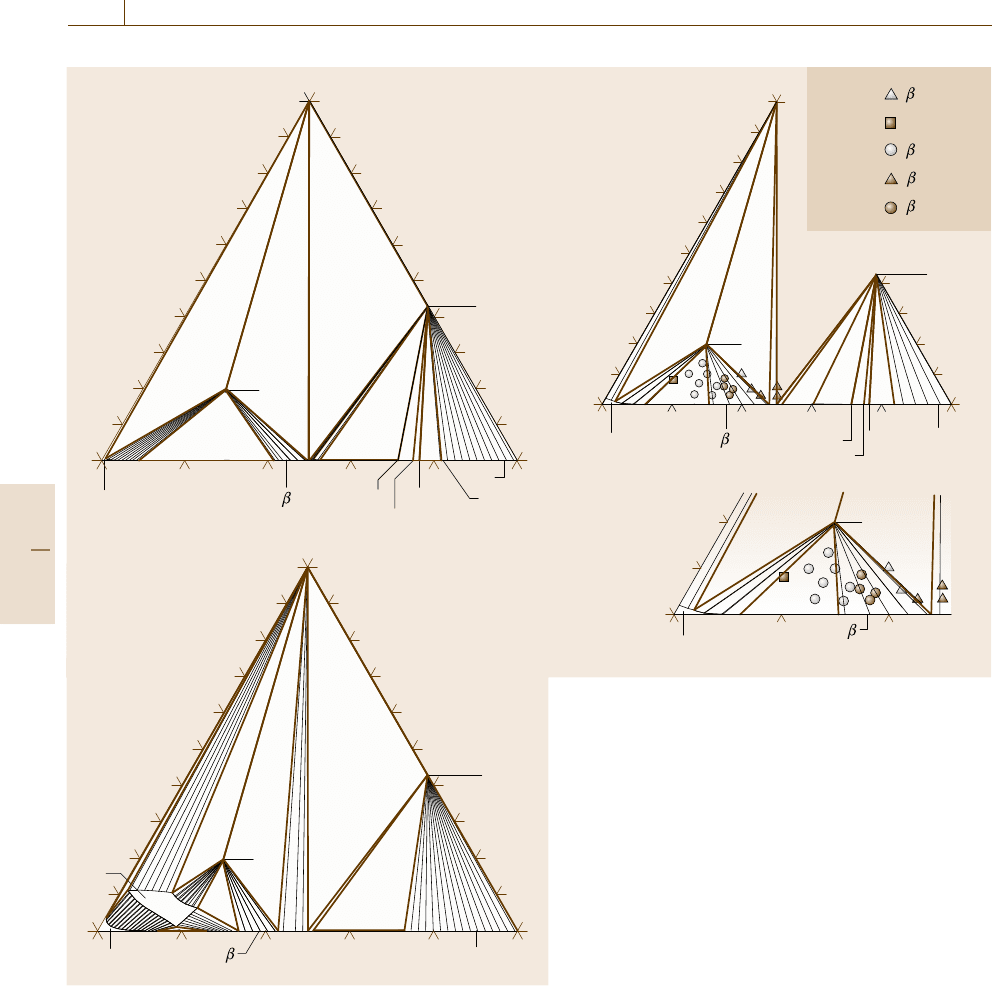
1086 Part E Modeling and Simulation Methods
10
20
30
40
50
60
70
80
90
a) 900 °C
10
20
30
40
50
60
70
80
90
C
20 40 60 80
Co Al
10
20
20 40
Co Al
b) 1100 °C
20
30
40
50
60
70
80
90
C
L
c)
10
20 40 60 80Co Al
1300 °C
γ(Co)
C content
(mol. %)
Co
20 40 60 80
AlL
Al4
3
C
3
Co
2
Al
5
CoAl
3
Co
4
Al
13
Co
2
Al
9
C
κ
Al content (mol. %)
γ(Co)
C content
(mol. %)
κ
Al content (mol. %)
Al
4
C
3
L
γ(Co)
C content
(mol. %)
γ
κ
Al content
(mol. %)
Kimura
et al.
Al
4
C
3
Co
2
A
5
Co
4
Al
13
L
CoAl
3
κ
Al (mol. %)
C (mol. %)
κ
+
+ (C)
γ
κ
+
+ (C)
+ γ
κ
+
κ
+
at a constant C content of 4 mol %, and an Al content
of 23 mol %, as shown in Fig. 20.29. In this figure, the
term κ
1
denotes the Ni-based L1
2
structure, while the
Co-based Perovskite structure is represented by κ
2
.At
higher temperatures, the E2
1
structure forms a homo-
geneous solution, and this phase is designated as κ.
Fig. 20.25a–c Isothermal section diagram of the Co
−
Al
−
C system calculated at: (a) 900
◦
C, (b) 1100
◦
C, and
(c) 1300
◦
C
The κ phase gradually changes its character from the
Co
3
AlC-based carbide to the Ni
3
Al-based intermetal-
lic compound with increasing Ni content in the alloy.
The precipitates decompose into almost stoichiomet-
ric Co
3
AlC and Ni
3
Al phases in the lower temperature
range.
Part E 20.3
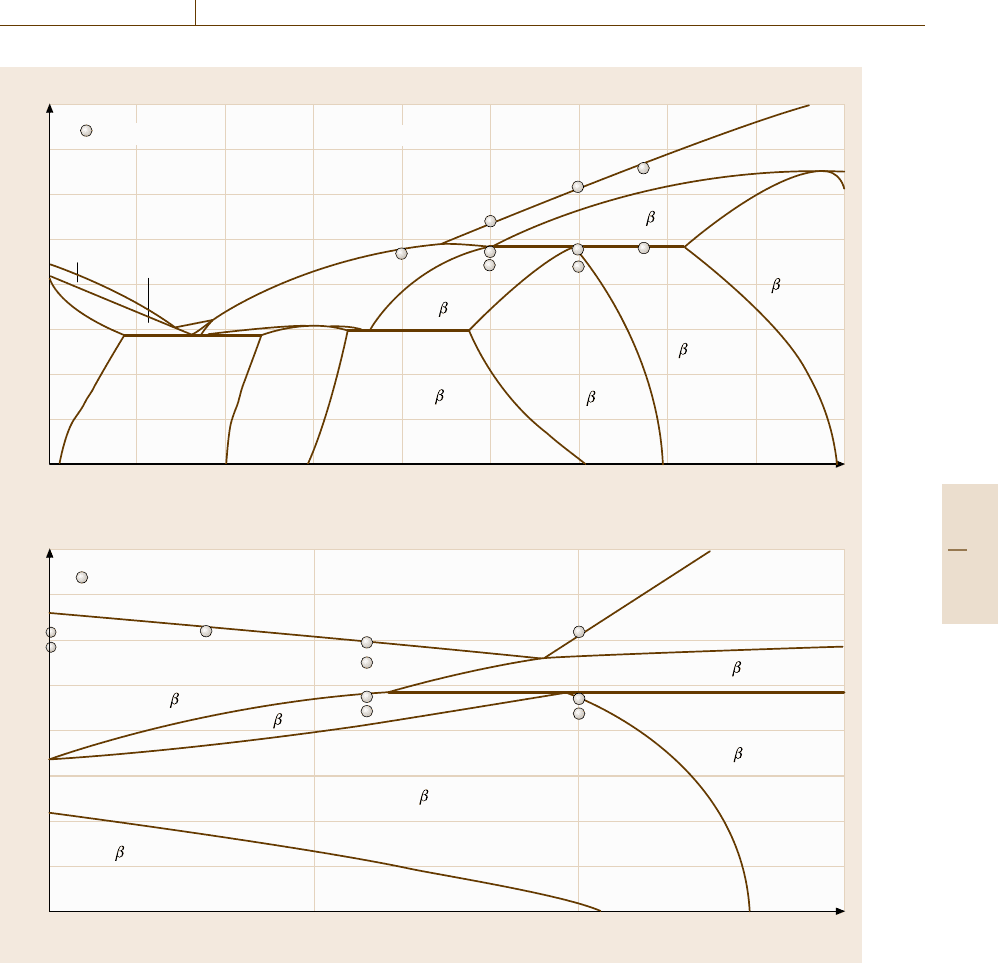
The CALPHAD Method 20.3 Prediction of Thermodynamic Properties of Compound Phases with First-principles Calculations 1087
1700
1600
1500
1400
1300
1200
1100
1000
900
Co
Al content (mol. %)
a) Temperature T (°C)
b) Temperature T (°C)
Co-Al-10 mol % C
90 mol. %
+ (C)
Kimura et al.
L
γ+L
γ+L
+ (C)γ
κ
+ +(C)
+L +(C)
L +(C)
+(C)
+(C)
γ
κ
+
κ
+L
+
κ
+L
γ +
κ
+
κ
+
1700
1600
1500
1400
1300
1200
1100
1000
900
Co 1510
455 10152025303540
5
C content (mol. %)
Co-30 mol % Al-C
70 mol. %
Kimura et al.
L
+
κ
+L
γ +
κ
+
κ
+
L + (C)
κ
+ + (C)
+L + (C)
+L
Fig. 20.26a,b Calculated vertical section diagrams at constant (a) 10 mol % C and (b) 30 mol % Al
Part E 20.3