Czichos H., Saito T., Smith L.E. (Eds.) Handbook of Metrology and Testing
Подождите немного. Документ загружается.

1058 Part E Modeling and Simulation Methods
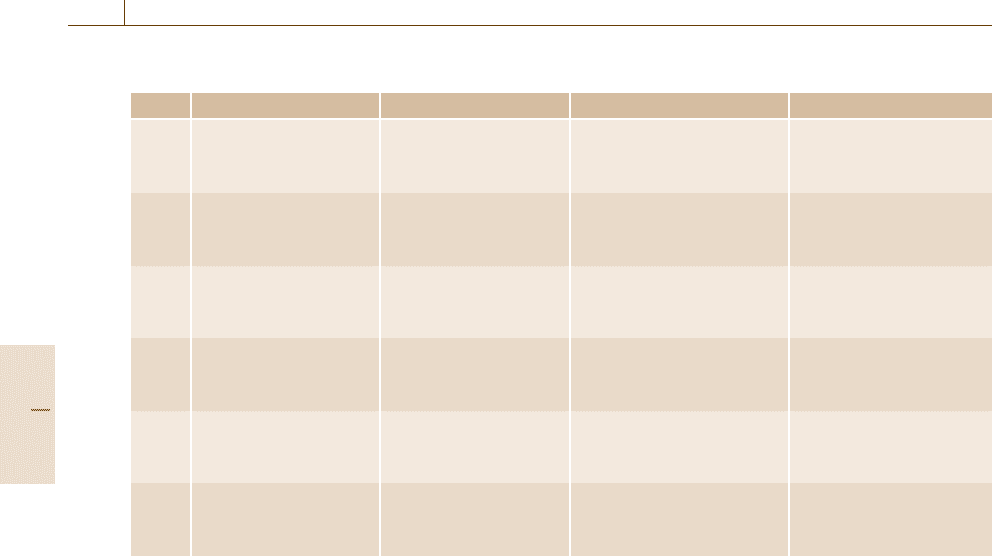
Table 19.1 Summary of discretized numerical schemes
Strong points Weak points Major targets Software
FEM 2nd BC treatment Calculation time Nonlinear solid/thermal/fluid/ ANSYS (multiphysics)
Local resolution Complex derivation vibration LS-DYNA (crash/solid)
Arbitrary shape Sensitive solution FIDAP (fluid), etc.
FDM Calculation time Stable solution Simple derivation Flow-3D (fluid), etc.
Stable solution Regular shape
Simple derivation Heuristic aspects
FVM Calculation time Local resolution Thermal/fluid FLUENT (fluid), etc.
Stable solution Heuristic aspects
Moving boundary
BEM Simple modeling Limited to linear/ Linear elastic/linear flow/ BEASY (solid), etc.
homogeneous case acoustic/magnetic
Dense matrix
CIP Complex problem New method Thermal/fluid/solid [19.17]
Stable solution No sales software
Multiphysics
PM No mesh/grid Calculation time Nonlinear solid/explosion [19.18]
Explosion New method
Highly nonlinear No sales software
greatly affects the calculation time and required mem-
ory size, some renumbering algorithms to reduce the
bandwidth are proposed.
The iterative methods can be classified into two
groups: (1) The stationary iterative methods, such as
the Jacobi method, Gauss–Seidel method, and suc-
cessive overrelaxation (SOR) method, and (2) the
nonstationary method, such as the conjugate gradi-
ent (CG) method, the biconjugate gradient stabilized
(Bi-CGSTAB) method, quasiminimal residual (QMR)
method, and generalized minimal residual (GMRES)
method [19.21]. The rough algorithm in the stationary
iterative methods can be represented by (19.126)
Ax =b → Mx =(M−A)x +b ,
Mx
k+1
=(M−A)x
k
+b , (19.126)
where (1) M = diagonal part of A,(2)M = triangular
part of A,and(3)M = combination of (1) and (2) part
of A.
The basic algorithm on nonstationary method is
represented in (19.127), where r, p,andx are the resid-
ual vector, direction vector, and the solution vector,
respectively.
r
0
=b−Ax
0
for k =0, 1, 2,...
p
k+1
= p
k
+αr
k
x
k+1
= x
k
+β p
k+1
r
k+1
=b−Ax
k+1
convergence check
end. (19.127)
To accelerate convergence of the iterations, the al-
gorithm with some preconditioners is proposed as
in [19.21]
r
0
=b−Ax
0
for k =0, 1, 2,...
z
k
=M
−1
r
i
p
k+1
= p
k
+αz
k
x
k+1
= x
k
+β p
k+1
r
k+1
=b− Ax
k+1
convergence check
end.
Some of the related subroutines above are available
from [19.22].
For the parallel calculation method such as the do-
main decomposition method, please refer to [19.23]
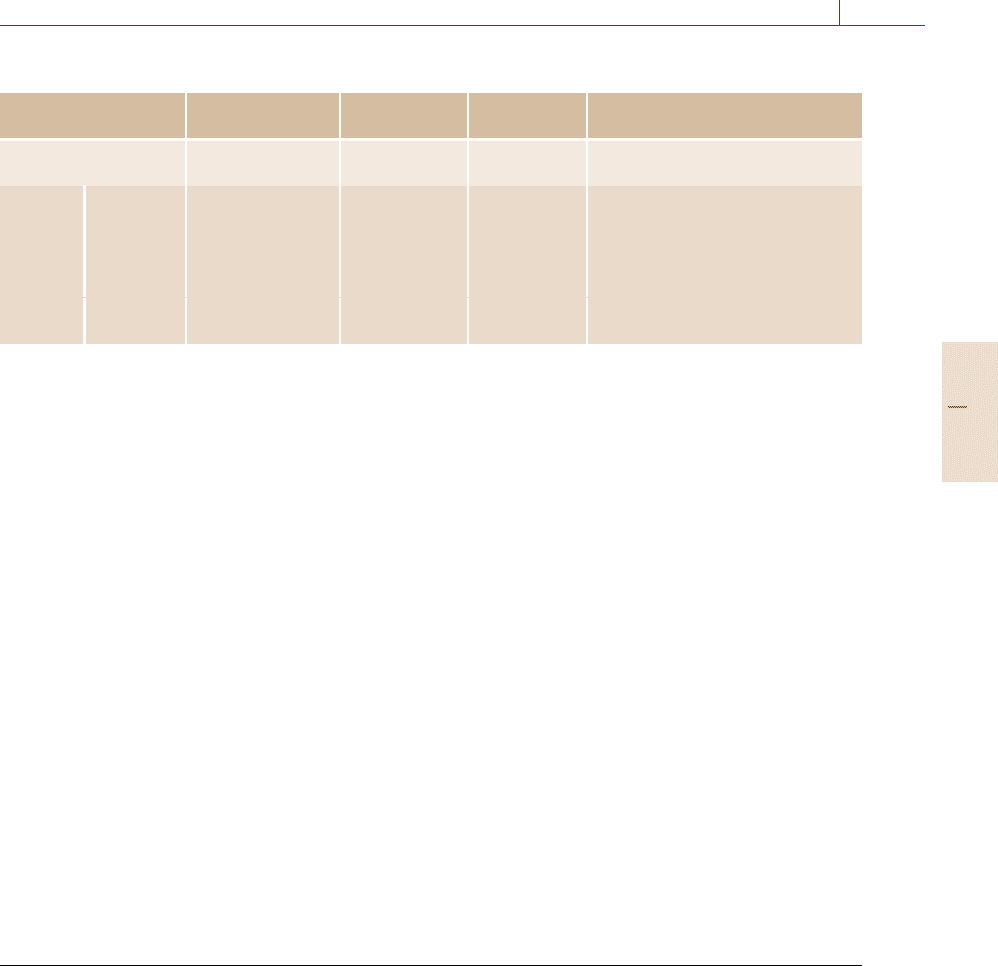
and [19.24]. Table 19.2 shows the summary of matrix
calculation methods.
Part E 19.9
Finite Element and Finite Difference Methods References 1059
Table 19.2 Summary of matrix calculation methods
For ill-conditioned For parallel Method
matrix calculation
Direct matrix solver ◦ × Skyline Symmetric
Frontal Nonsymmetric
Iterative Stationary × ◦ Jacobi M =diagonal part of A
matrix Gauss–Seidel M =triangular part of A
solver SOR M =diagonal and triangular parts of A
Non- × ◦ CG Symmetric/FEM/solid
stationary Bi-CGSTAB Nonsymmetric/FEM/solid
QMR Nonsymmetric/FEM/fluid
GMRES Nonsymmetric/FEM/fluid
19.9.6 Multiscale Method
To bridge the macroscopic scale and mezoscopic scale,
the multiscale approach was recently considered to be
important. The homogenization method is one of the so-
lutions that carries numerical material testing between
macroscale (usual FE mesh) and microscale (FE mesh
at material level) to get real-time stress–strain constitu-
tive behavior for FEA in macroscale. The key points of
this method are introduced in reference [19.25].
19.10 Free Codes
Some internet URLs for FEM and FDM are provided
for the reader’s convenience. This is not a recommended
selection; the reader must use the information at their
own risk.
1. Information on modeling, FEM,andFDM
http://homepage.usask.ca/∼ijm451/finite/fe_
resources/fe_resources.html (Internet Finite Ele-
ment Resources; general information on FEM
including free codes)
http://morden.csee.usf.edu/dragon/kpalbrec/mesh.
html (Meshing Research Corner; survey of mesh
generation schemes and codes)
2. Parallel computations in FEM, FDM,andFVM
http://www.ibase.aist.go.jp/infobase/pcp/platform-
en/index.html (Parallel Computing Platform/PCP
developed by AIST and FUJIRIC; perfect solution
for parallelization of FEM/FDM/FVM codes, free
from headaches from matrix handling, MPI com-
mands and memory allocations, sample programs of
FEM/FDM/FVM are also provided)
3. Related libraries
http://www.netlib.org/ (collection of mathematical
software, papers, and databases)
4. Modeling-related free software
http://www.netlib.org/voronoi/index.html (Voronoi;
2-dimensional Voronoi diagram, Delaunay triangu-
lation)
http://www.geuz.org/gmsh/ (Gmsh mesh generation
code with TRI/QUAD/TETRA/HEXA element)
References
19.1 J.W. Thomas: Numerical Partial Differential Equa-
tions: Finite Difference Methods (Springer, Berlin,
Heidelberg 1995)
19.2 T.J.R. Hughes: The Finite Element Method: Linear
Static and Dynamic Finite Element Analysis (Dover,
New York 2000)
19.3 A.N. Kolmogorov, S.V. Fomin: Elements of the The-
ory of Functions and Functional Analysis (Dover,
New York 1999)
19.4 G. Strang: Introduction to Applied Mathematics
(Wellesley-Cambridge, Cambridge 1986)
19.5 N. Kikuchi: Finite Element Methods in Mechanics
(Cambridge Univ. Press, New York 1986)
19.6 T.Belytschko,W.K.Liu,B.Moran:Nonlinear Finite
Elements for Continua and Structures (Wiley, New
York 2000)
19.7 S.P. Timoshenko, J.N. Goodier: Theory of Elasticity
(McGraw-Hill, New York 1970)
Part E 19

1060 Part E Modeling and Simulation Methods
19.8 O.C. Zienkiewicz, D.V. Phillips: An automatic mesh
generation scheme for plane and curved surfaces
by isoparametric coordinate, Int. J. Numer. Meth-
ods Eng. 3, 519–528 (1971)
19.9 J.F. Thompson, Z.U.A. Warsi, C.W. Mastin: Numer-
ical Grid Generation (North-Holland, Amsterdam
1985)
19.10 A. Tezuka: 2D mesh generation scheme for adaptive
remeshing process, Jpn. Soc. Mech. Eng. 38,204–
215 (1996)
19.11 A.M. Winslow: Numerical solution of the quasi-
linear poisson equation in a nonuniform triangle
mesh, J. Comput. Phys. 2, 149–172 (1967)
19.12 M. Ainsworth, J.T. Oden: A Posterior Error Estima-
tion in Finite Element Analysis (Wiley, New York
2000)
19.13 P.J. Roache: Verification and Validation in Com-
putational Science and Engineering (Hermosa,
Socorro 1998)
19.14 R. Peyret: Handbook of Computational Fluid Me-
chanics (Academic, New York 1996)
19.15 K.A. Woodbury: Inverse Engineering Handbook
(CRC, Boca Raton 2002)
19.16 J.H. Ferziger, M. Peric: Computational Methods for
Fluid Dynamics (Springer, Berlin, Heidelberg 2001)
19.17 C.A. Brebbica: The Boundary Element Method for
Engineers (Pentech, London 1978)
19.18 T. Nakamura, R. Tanaka, T. Yabe, K. Takizawa:
Exactly conservative semi-Lagrangian scheme for
multidimensional hyperbolic equations with di-
rectional splitting technique, J. Comput. Phys. 174,
171–207 (2001)
19.19 CIPUS (CIP User’s Society): www.mech.titech.ac.jp/
~ryuutai/cipuse.html (2011) by T. Yabe
19.20 W.K. Liu, S. Li: Meshfree and particle methods
and their applications, Appl. Mech. Rev. 55,1–34
(2002)
19.21 G.H. Golub, C.F. Van Loan: Matrix Computations
(Johns Hopkins Univ. Press, Baltimore 1996)
19.22 W.H. Press, S.A. Teukolsky, W.T. Vetterling,
B.P. Flannery: Numerical Recipes in Fortran 90:
The Art of Parallel Scientific Computing, 2nd edn.
(Cambridge Univ. Press, Cambridge 1996)
19.23 B.F. Smith, P.E. Bjrstad, W.D. Gropp: Domain De-
composition: Parallel Multilevel Methods for Elliptic
Partial Differential Equations (Cambridge Univ.
Press, Cambridge 1996)
19.24 Parallel Computing Platform/PCP: www.ibase.aist.
go.jp/infobase/pcp/platform-en/index.html (2011)
19.25 J.M. Guedes, N. Kikuchi: Preprocessing and post-
processing for materials based on the homog-
enization method with adaptive finite element
methods, Comput. Methods Appl. Mech. Eng. 83,
143–198 (1990)
Part E 19
1061
The CALPHAD
20. The CALPHAD Method
Phase diagrams offer various areas of materials
science and technology indispensable informa-
tion for the comprehension of the properties of
materials. The microstructure of solid materials is
generally classified according to the size of the con-
stituents – for example, at the electron, atomic, or
granular level (Sect. 1.3). Accordingly, fundamen-
tal principles like quantum mechanics, statistical
mechanics, or thermodynamics are applied indi-
vidually to describe the physical properties. Phases
are important features of material because they
characterize homogeneous aggregations of matter
with respect to chemical composition and uniform
crystal structure. The various functions of a mater-
ial are closely related to the phases and structures
of the material’s composition. Therefore, to de-
velop a material with a maximum level of desired
functions, it is essential to undertake design of the
structure in advance.
Phase diagrams are composed by means of
experimental measurements, as well as statisti-
cal thermodynamic analysis. The construction of
phase diagram calculations based on experiments
and thermodynamic analysis are generally referred
to as the calculation of phase diagrams (CALPHAD)
approach [20.1]. This method provides a very ac-
curate understanding of the properties originating
in the macroscopic character of the material under
study.
This chapter is organized in three parts:
•
In the first part, a brief outline of the CALPHAD
method is summarized.
•
In the second part, the method for deriving the
Gibbs free energies incorporating the ab initio
calculations is presented in order to clarify the
uncertainty of thermodynamic properties for
metastable solution phases, taking the Fe
−
Be-
based bcc phase as an example. Some results
20.1 Outline of the CALPHAD Method..............1062
20.1.1 Description of Gibbs Energy ...........1062
20.1.2 Equilibrium Conditions .................1064
20.1.3 Evaluation
of Thermodynamic Parameters.......1065
20.2 Incorporation of the First-principles
Calculations into the CALPHAD Approach . 1066
20.2.1 Outline
of the First-principles Calculations . 1066
20.2.2 Gibbs Energies of Solution Phases
Derived by the First-principles
Calculations.................................1067
20.2.3 Thermodynamic Analysis
of the Gibbs Energies Based
on the First-principles Calculations 1071
20.2.4 Construction of Stable
and Metastable Phase Diagrams.....1071
20.2.5 Application
to More Complex Cases..................1073
20.3 Prediction of Thermodynamic Properties
of Compound Phases with
First-principles Calculations ..................1079
20.3.1 Thermodynamic Analysis
of the Fe–Al–C System ..................1079
20.3.2 Thermodynamic Analysis
of the Co–Al–C and Ni–Al–C
Systems ......................................1083
References ..................................................1090
for metastable phase equilibria in the Fe
−
Be,
and Co
−
Al binary systems are shown.
•
In the third part the application to predict ther-
modynamic properties of compound phases is
discussed. The thermodynamic modeling for
thePerovskitecarbidewithanE2
1
-type struc-
ture in the Fe
−
Al
−
C, Co
−
Al
−
C and Ni
−
Al
−
C
ternary systems is illustrated, and constructions
of phase diagrams are performed.
Part E 20
1062 Part E Modeling and Simulation Methods
20.1 Outline of the CALPHAD Method
Phase diagrams provide basic and important infor-
mation especially for the design of new materials.
However, by using experimental techniques only, a lot
of time and labor is required in construction of even
a partial region of a phase diagram, because the practical
materials are composed of multicomponent alloys, more
than ternary systems. In order to break this difficult situ-
ation, the method of calculation of phase diagrams was
advocated and is referred to as the CALPHAD method.
CALPHAD was originally a name for this researcher’s
group, however it has recently become the name for the
technique by which a phase diagram is calculated on the
basis of thermodynamics. In this method a variety of ex-
perimental values concerning the phase boundaries and
the thermodynamic properties is analyzed according to
an appropriate thermodynamic model and the interac-
tion energies between atoms are evaluated. By using
this technique, phase diagrams outside the experimen-
tal range can be calculated based on thermodynamic
proof. Difficulty in extension of the calculated results
to higher-order systems is much less than that in the
case of experimental work, since the essence of the cal-
culation does not change so much between a binary
system and a higher-order system. This method pro-
vides a very accurate understanding of the properties
originating in the macroscopic character of the material
under study. However, a shortcoming of this approach
is that it is hard to obtain information on metastable
equilibria, or on undiscovered phases, since the ther-
modynamic parameters from this method can only be
evaluated from experimental data.
The empirical model of de Boer et al. [20.2]hasof-
ten been used to deduce the thermodynamic quantities
of systems where the experimental values do not exist.
In this model, generally called Miedema’s model, the
crystal lattice is divided into Wigner–Seitz cells. The
simple expression for the formation energy in alloys is
derived by considering the change in the electron den-
sity states at the interface between the cells, as shown
in (20.1)
ΔE ∝−P
(
ΔΦ
)
2
+Q
Δn
1/3
WS
2
. (20.1)
Here, Δn
WS
is the difference in electron density based
on the volume of the Wigner–Seitz cell between differ-
ent species of atoms. This term always leads to local
perturbations that give rise to a positive energy con-
tribution in (20.1). On the other hand, ΔΦ presents
a difference between the chemical potentials of the dif-
ferent species of atoms at the cell surfaces, and leads to
an attractive term in (20.1). The constants P and Q are
proportionality constants, derived empirically. Equa-
tion (20.1) does not contain any parameters that refer
to the crystal structure, and therefore, the heats of solu-
tion of face-centered-cubic (fcc), body-centered-cubic
(bcc), and hexagonal-close-packed (hcp) structures, as
well as liquids, are predicted. However, it is known that
the absolute values are not always predicted accurately,
although this model predicts the correct sign for the for-
mation enthalpy in various alloy systems. For example,
thermodynamic analysis of the Fe
−
Pd system predicts
the enthalpy of formation for the L1
0
and the L1
2
ordered phases to be −14 kJ/mol and −22 kJ/mol, re-
spectively, while the predicted values from Miedema’s
model are −6kJ/mol for the L1
0
phase and −4kJ/mol
for the L1
2
phase [20.3]. The large difference between
these values means that incorporating Miedema’s model
with a thermodynamic analysis is rather difficult.
Such serious problems, which are intrinsic to the
CALPHAD approach, should be solved with the as-
sistance of the first-principles energetic calculation
method. Thus, in the present chapter, a new approach
to introduce some thermodynamic quantities obtained
by the ab initio band energy calculations in the conven-
tional CALPHAD-type analysis of some alloy systems
is presented.
20.1.1 Description of Gibbs Energy
Gibbs Free Energy
Using the Regular Solution Approximation
The selection of a thermodynamic model by which
Gibbs free energy of an alloy system is described is
the most important factor when using the CALPHAD
method. In a system in which interaction between alloy-
ing elements is not so strong, the regular solution model
is well known to describe the thermodynamic properties
of the alloys comparatively well. For instance, the free
energy of the A
−
B binary alloy is expressed as (20.2)
G
m
= x
A
0
G
A
+x
B
0
G
B
+x
A
x
B
L
AB
+RT(x
A
ln x
A
+x
B
ln x
B
) , (20.2)
where
0
G
i
is the Gibbs energy of the pure component i
in the standard, and x
i
is the mole fraction of i. L
AB
shows the interaction energy for A atoms and B atoms,
and is given by
L
AB
= NZ
ε
AB
−
1
2
(
ε
AA
+ε
BB
)
, (20.3)
Part E 20.1
The CALPHAD Method 20.1 Outline of the CALPHAD Method 1063
where N denotes the so called Avogadro’s number, Z is
the coordination number, and ε
AB
, ε
AA
and ε
BB
show
the cohesion energies between A–B, A–A, B–B atomic
pair, respectively. The ordering between unlike atoms
occurs for L
AB
< 0 because the A–B atom pair is more
stable than the average of the A–A and B–B atom pairs.
On the other hand, the clustering of the like atom is
caused in case of L
AB
> 0. For L
AB
= 0, no atomic
interaction exists, and then the solution shows the ran-
dom mixing of atoms. This interaction parameter should
not be constant but a function of the temperature and
the chemical compositions of alloys. From a qualita-
tive point of view, the first term of (20.2) represents the
energies of mechanical mixing, the second term stands
for excess energy which shows deviation from the ideal
solution, and the third is ideal mixing entropy.
Gibbs Free Energy Using the Sublattice Model
For the simplest case of the sublattice model [20.4],
let us consider a phase represented by the formula
(A, B)
a
(C, D)
b
, containing two elements A and B in one
sublattice “1” and two elements C and D in the other
sublattice “2”. The coefficients a and b show the number
of sites in each sublattice and the size of the sublattices
may be generally chosen as a +b =1 for convenience.
There are four elements in the system, however the de-
grees of freedom in varying in composition are two,
because the total numbers of atoms in each sublat-
tice are fixed. Thus these two composition parameters
are conveniently plotted on a square where the corners
A
a
D
b
y
D
2
y
B
1
B
a
D
b
B
a
C
b
A
a
C
b
0
G
A
a
D
b
0
G
A
a
C
b
0
G
B
a
C
b
0
G
B
a
D
b
a)
b)
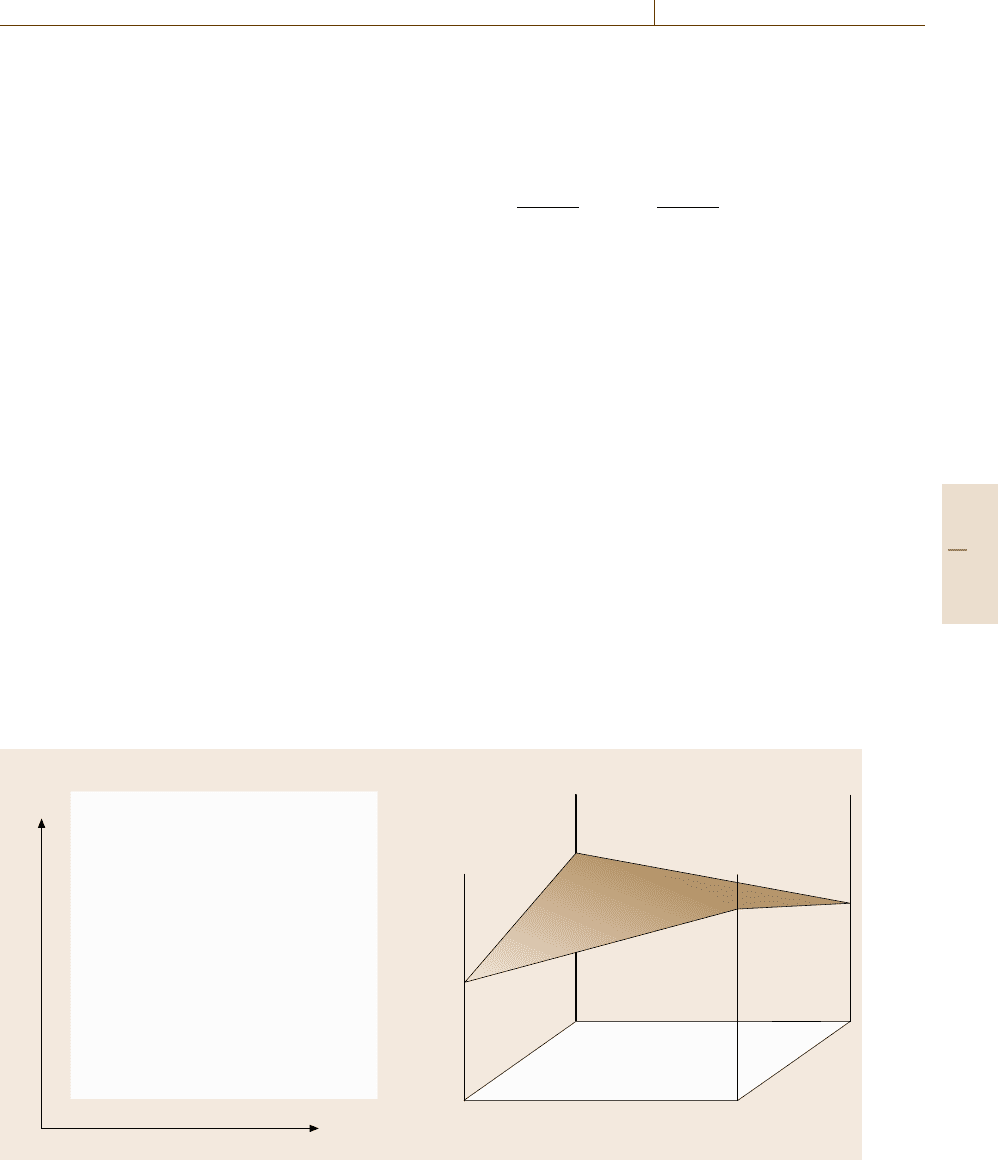
Fig. 20.1 (a) Composition square in a quaternary system (A, B)
a
(C, D)
b
and (b) surface of reference for the free energy
represented the four basic end members A
a
C
b
,A
a
D
b
,
B
a
C
b
,andB
a
D
b
, as shown in Fig. 20.1a. The symbols
y
1
B
and y
2
D
denote the mole fractions, respectively, for
the sublattices 1 and 2, and they are represented as fol-
lows:
y
1
B
=
n
1
B
n
1
A
+n
1
B
, y
2
D
=
n
2
D
n
2
C
+n
2
D
. (20.4)
In this equation n
j
i
denotes the number of i atoms in
the j sublattice. The nonplanar surface composed of
four reference states, i. e., one each for the compounds
A
a
C
b
,A
a
D
b
,B
a
C
b
,andB
a
D
b
, is called the surface of
reference as shown in Fig. 20.1b, and represented by
G
ref
m
= y
1
A
y
2
C
0
G
A
a
C
b
+y
1
A
y
2
D
0
G
A
a
D
b
+y
1
B
y
2
C
0
G
B
a
C
b
+y
1
B
y
2
D
0
G
B
a
D
b
. (20.5)
For the entropy term, the following Gibbs energy of
mixing can be obtained, assuming the atoms mix ran-
domly within each sublattice
G
ideal
mix
= RT
a
y
1
A
ln y
1
A
+y
1
B
ln y
1
B
+b
y
2
C
ln y
2
C
+y
2
D
ln y
2
D
. (20.6)
It seems difficult to accept that the interaction between
A and B atoms should be quite independent of whether
the other sublattice is occupied by C and D atoms. Thus
the regular solution model should be defined as the
power series representation of excess Gibbs free energy
Part E 20.1

1064 Part E Modeling and Simulation Methods
by the following equation
G
xs
mix
= y
1
A
y
1
B
y
2
C
0
L
A,B:C
+y
1
A
y
1
B
y
2
D
0
L
A,B:D
+y
2
C
y
2
D
y
1
A
0
L
A:C,D
+y
2
C
y
2
D
y
1
B
0
L
B:C,D
,
(20.7)
where L
i, j:k
(or L
i:j,k
) is the interaction parameter
between unlike atoms on the same sublattice. Conse-
quently the Gibbs energy for one mole of the phase,
denoted as (A, B)
a
(C, D)
b
, can be represented by the
two-sublattice model as shown in (20.8):
G
m
= G
ref
m
+G
ideal
mix
+G
xs
mix
. (20.8)
Gibbs Energy for Interstitial Solutions
Represented by Sublattice Model
The Gibbs energy for interstitial phases is very impor-
tant in steels and ferrous alloys, where the elements
such as C, N, S, or B occupy the interstitial sites of
solid solutions. The structure of such a phase is consid-
ered as consisting of two sublattices, one occupied by
substitutional elements and the other occupied by the
interstitial elements as well as vacancies. Let us con-
sider the austenite phase for the Fe
−
Cr
−
C system, for
instance. The occupation of the sublattices is shown as
(Cr, Fe)
1
(C, Va)
1
, since the number of sites in each sub-
lattice is 1 :1 in the case of an fcc structure. The Gibbs
energy is given by the following equation:
G
m
= y
1
Cr
y
2
Va
0
G
Cr:Va
+y
1
Fe
y
2
Va
0
G
Fe:Va
+y
1
Cr
y
2
C
0
G
Cr:C
+y
1
Fe
y
2
C
0
G
Fe:C
+RT
y
1
Cr
ln y
1
Cr
+y
1
Fe
ln y
1
Fe
+
y
2
C
ln y
2
C
+y
2
Va
ln y
2
Va
+y
1
Cr
y
1
Fe
y
2
Va
v
v
L
Cr,Fe:Va
y
1
Cr
−y
1
Fe
v
+y
1
Cr
y
1
Fe
y
2
C
v
v
L
Cr,Fe:C
y
1
Cr
−y
1
Fe
v
+y
1
Cr
y
2
C
y
2
Va
v
v
L
Cr:C,Va
y
2
C
−y
2
Va
v
+y
1
Fe
y
2
C
y
2
Va
v
v
L
Fe:C,Va
y
2
C
−y
2
Va
v
.
(20.9)
The first two terms represent the Gibbs energy of fcc Cr
and Fe, because the second sublattice consists of vacan-
cies. The next two terms represent the Gibbs energy of
the CrC and FeC compounds, where the interstitial sites
are completely filled with C. The second line shows the
ideal entropy term, while the residual lines represents
the excess terms of the Gibbs energy. When y
2
C
= 0in
the equation, the Gibbs energy equation coincides with
the equation for the simple substitutional mixing of Cr
and Fe.
Magnetic Contribution to Gibbs Energy
In magnetic materials containing Fe, Ni and Co, there
is polarization of electron spins, and it is necessary to
consider the magnetic contribution to the Gibbs energy,
by adding the extra term to the paramagnetic Gibbs
energy G
m
as follows:
G = G
m
+G
mag
. (20.10)
The magnetic contribution to the Gibbs free energy,
G
mag
, is given by the expression
G
mag
= RTf (τ)ln(β +1) , (20.11)
where
f (τ) =1−
1
A
79τ
−1
140p
+
474
497
1
p
−1
×
τ
3
6
+
τ
9
135
+
τ
15
600
, for τ<1
(20.12)
and
f (τ) =−
1
A
τ
−5
10
+
τ
−15
315
+
τ
−25
1500
, for τ ≥1 ,
(20.13)
and
A =
518
1125
+
11 692
15 975
1
p
−1
. (20.14)
The variable τ is defined as T/T
C
, where T
C
is the
Curie temperature and β is the mean atomic moment ex-
pressed in Bohr magnetons μ
B
. The value of p depends
on the structure, and p =0.28 for the fcc phase.
20.1.2 Equilibrium Conditions
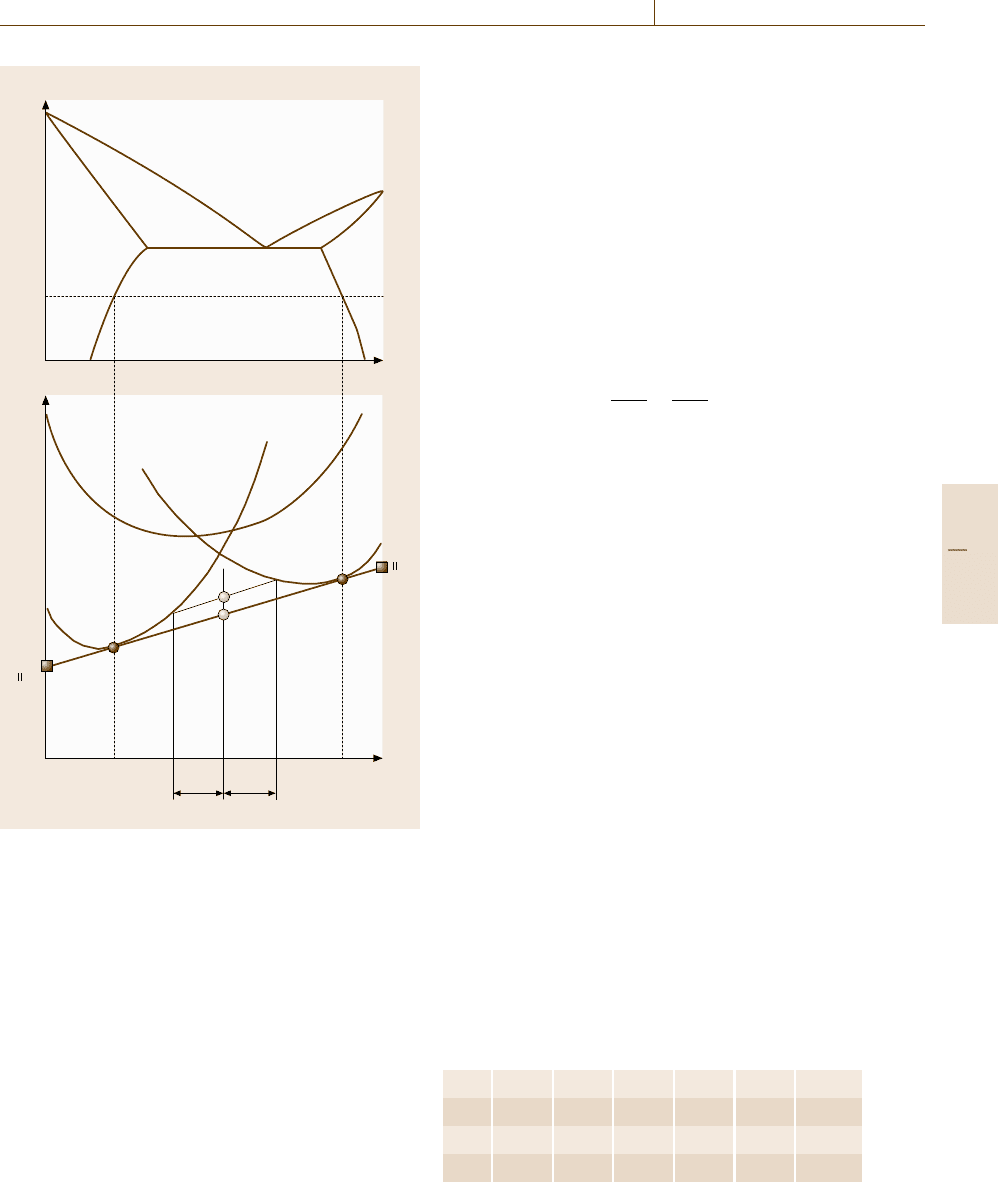
Figure 20.2 shows the so-called Gibbs energy-
composition diagram for the A
−
B binary system. This
figure schematically shows the relationship between
a eutectic type of binary phase diagram and the Gibbs
energy curves of each phase. Let us consider the case in
which the microstructure of the alloy is composed of the
Part E 20.1
The CALPHAD Method 20.1 Outline of the CALPHAD Method 1065
T
1
Temperature
Liq
α
β
Gibbs energy
G
Liq
G
β
G
α
Content
a
1
a
e
G
e
G
1
b
1
b
e
ν
A
α
x
B
β
f
β
f
α
B
α
x
AB
B
x
1
β
ν
A
ν
B
α
β
ν
B
Fig. 20.2 Eutectic type of phase diagram and correspond-
ing Gibbs energy-composition diagram for hypothetical
A
−
B binary system
α and β phases. When the Gibbs energies of the α and
β phases are given by the points a
1
G
α
1
and b
1
G
β
1
, re-
spectively, the total energy of the solution is represented
by the line a
1
b
1
in the figure. At the alloy composition
x
1
B
, the Gibbs energy is G
1
and each phase has the vol-
ume fraction of f
α
and f
β
. Then the following relation
holds:
G
1
= G
α
1
f
α
+G
β
1
f
β
. (20.15)
The thermodynamic equilibrium is achieved under the
condition of the lowest energy of the alloy. According to
this criterion, the point G
1
is not the lowest energy state,
but decreases further to G
e
by changing f
α
and f
β
.
The line a
e
b
e
is a common tangent of Gibbs energy G
α
and G
β
, and the equilibrium composition of each phase
is given as x
α
B
and x
β
B
. In the calculation of the phase
equilibrium, it is convenient to use these chemical po-
tentials as the basic equations. For instance, to obtain
the equilibrium between phase α and phase β,(20.16)
should be calculated by using the numerical analytic
method
μ
α
A
=μ
β
A
,
μ
α
B
=μ
β
B
. (20.16)
The chemical potential μ of each element is obtained by
using
μ
i
= G
m
−
2
j=1
x
j
∂G
m
∂x
j
+
∂G
m
∂x
i
(20.17)
and hence the chemical potentials of A and B species in
the φ phase are expressed as:
μ
φ
A
=
0
G
φ
A
+RT ln x
φ
A
+
x
φ
B
2
L
φ
AB
,
μ
φ
B
=
0
G
φ
B
+RT ln x
φ
B
+
x
φ
A
2
L
φ
AB
. (20.18)
20.1.3 Evaluation
of Thermodynamic Parameters
The interaction parameters included in the free energy
expression can be directly evaluated from the experi-
mental values on activity. For example the activity of
Pb over the Pb
−
Sb binary liquid have been determined
asshowninTable20.1 [20.5]. The liquid state of the
elements is adopted as its standard. If the composition
dependency of the interaction parameter is expressed
as (20.19), the chemical potential of Pb in the binary
system can be derived as (20.20)
L
PbSb
=
0
L
PbSb
+
x
Pb
−x
Sb
1
L
PbSb
, (20.19)
μ
Pb
=
0
G
Pb
+RT ln x
Pb
+
1−x
Pb
2
×
0
L
PbSb
+
4x
Pb
−1
1
L
PbSb
. (20.20)
Table 20.1 Experimental data on activity of Pb in the
Pb
−
Sb binary liquid
x
Pb
0.05 0.1 0.2 0.3 0.4 0.5
a
Pb
0.04 0.08 0.162 0.247 0.338 0.433
x
Pb
0.6 0.7 0.8 0.9 0.95
a
Pb
0.555 0.674 0.785 0.897 0.946
Part E 20.1
1066 Part E Modeling and Simulation Methods
This is straightforwardly arranged as
RT ln
a
Pb
/x
Pb
1 −x
Pb
2
=
0
L
PbSb
−
1
L
PbSb
+4x
Pb
1
L
PbSb
(20.21)
since the relation μ
Pb
=
0
G
Pb
+RT ln a
Pb
holds. There-
fore, if the left-hand side of (20.21) is calculated from
the activity data shown in Table 20.1 and the each value
is plotted against x
Pb
, the interaction parameters
0
L
PbSb
,
1
L
PbSb
can be obtained, respectively, from the intercept
of the ordinate and the slope of the straight line as:
0
L
PbSb
=−2900 J/mol ,
1
L
PbSb
=−200 J/mol .
(20.22)
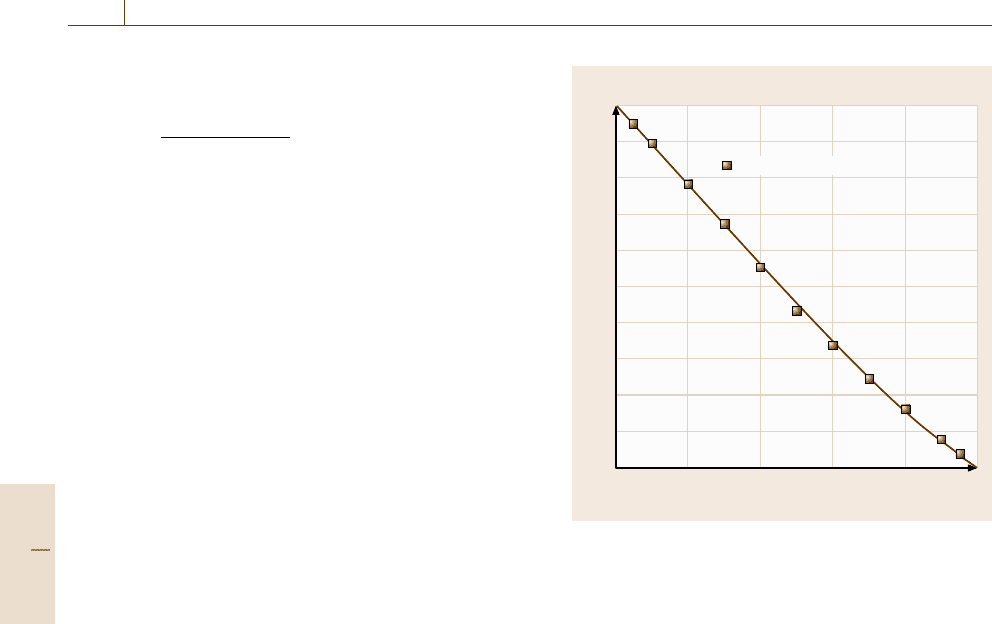
The calculated activity of Pb in the liquid phase
was compared with the experimental values in Fig. 20.3.
Considering the other observed data set at a different
temperature yields the dependence on the temperature
for the interaction parameters
0
L
PbSb
,
1
L
PbSb
.
1
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0
0 0.2 0.4 0.6 0.8 1
Sb/atom fraction
Activity of Pb
Oelsen et al. (903 K)
Fig. 20.3 Calculated activity of Pb in the Pb
−
Sb binary
liquid compared with experimental values [20.5]
20.2 Incorporation of the First-principles Calculations
into the CALPHAD Approach
In this section, in order to clarify the uncertainty
of thermodynamic properties for metastable solution
phases, the method for deriving the Gibbs free en-
ergies incorporating the first-principles calculations is
presented, taking the Fe
−
Be-based bcc phase as the
example. First, a set of some superstructures is se-
lected to be representative of a series of bcc-based
ordered phases, and the total energies are calculated
using the first-principles calculations. Next, the effec-
tive cluster interaction energies can be extracted from
these formation energies using the cluster expansion
method (CEM). This leads to a set of composition-
independent parameters, from which the energy of
the set of superstructures can be reproduced in terms
of a set of correlation functions. Once we know the
cluster interaction energies for the alloy system, the
enthalpy term is expressed by the combination of ef-
fective cluster interaction and the correlation functions
for the clusters. The entropy term in the Gibbs en-
ergy expression is calculated using the cluster variation
method (CVM) with the tetrahedron approximation to
calculate the configurational entropy. Minimizing the
grand potential with respect to all the correlation func-
tions allows for the Gibbs energy of mixing to be
obtained as a function of composition at a constant
temperature.
20.2.1 Outline
of the First-principles Calculations
First-principles calculations using density functional
theory (DFT) have proved to be quite reliable in con-
densed matter physics. There still remains a barrier to
overcome in application to materials science, for in-
stance, stoichiometric deviations, surfaces, impurities,
and grain boundaries, however, for certain materials, the
direct application of this technique has been attempted
in studying the various properties using 100 or more
atoms in a unit cell. According to the theorem based
on DFT, the total energy E
tot
of a nonspin-polarized
system of interacting electrons in an external poten-
tial is given as a function of the ground state electronic
Part E 20.2
The CALPHAD Method 20.2 Incorporation of the First-principles Calculations into the CALPHAD Approach 1067
density ρ as in the following equation:
E
tot
(ρ) =
V(r)ρ(r)d
3
r +T[ρ]
+
e
2
2
ρ(r)ρ(r
)
|r −r
|
d
3
r d
3
r
+E
XC
[ρ].
(20.23)
Here the first term
V(r)ρ(r)d
3
r denotes the Coulomb
interaction energy between the electrons and nu-
clei, T[ρ] is the single-particle kinetic energy,
e
2
2
ρ(r)ρ(r
)
|r−r
|
d
3
r d
3
r
is the Hartree component of the
electron–electron energy, and E
XC
[ρ] is the exchange-
correlation functional.
Kohn and Sham [20.6] showed that the correct
density in the equation is given by the self-consistent so-
lution of a set of single particles following Schrödinger-
like equations as
ε
i
Ψ
i
(r) =
−
2m
∇
2
+V(r) +e
2
×
ρ(r
)
|r −r
|
d
3
r
+V
XC
(r)
Ψ
i
(r) .
(20.24)
In (20.24), V
XC
represents the exchange correlation
potential and V
XC
(r) =∂E
XC
[ρ]/∂ρ(r). Thus we can
obtain an appropriate electronic density in much eas-
ier way as compared with solving a many-body
Schrödinger equation. In calculations, the single par-
ticle Kohn–Sham equation in (20.24) is solved sepa-
rately on a grid of sampling points in the irreducible
part of the Brillouin zone, and the obtained orbitals are
used to construct the charge density.
The first-principles calculations based on DFT may
be, at this time, divided into two methods; one employ-
ing pseudopotentials and relatively simple basis sets,
Table 20.2 Comparison of the estimated formation enthalpies for some ordered structures with the appropriate evaluated
data
Alloy Structures Temperature Experimental values Calculated values
systems (K) (kJ/mol) (kJ/mol)
Al
−
Ni B2 298 −71.7 −69.5
L1
2
298 −41.0 −40.5
Al
−
Ti L1
0
298 −39.8 −39.6
D0
19
(Ti
3
Al) 298 −27.5 −27.3
Be
−
Cu B2 298 −21.4 −19.0
Fe
−
Pd B2 298 −15.6 −13.2
Fe
−
Ti B2 298 −51.6 −46.5
Ni
−
Ti B2 999 −34.2 −34.1
D0
24
(Ni
3
Ti) 827 −54.0 −51.4
Al
−
Li B32 298 −22.8 −23.1
and the other using much more complex basis sets such
as the full potential linearized augmented plane wave
(FLAPW), which gives accurate results on formation
energies for metals. The FLAPW method, as embodied
in the WIEN2k software package [20.7], is one of the
most accurate schemes for electronic calculations, and
allows for very precise calculations of the total energies
in a solid, and will be employed in the present ener-
getic calculations. The FLAPW method uses a scheme
for solving many-electron problems based on the lo-
cal spin density approximation (LSDA) technique. In
this framework, a unit cell is divided into two regions:
nonoverlapping atomic spheres and an interstitial re-
gion. Inside the atomic spheres, the wave functions of
the valence states are expanded using a linear combina-
tion of radial functions and spherical harmonics, while
a plane wave expansion is used in the interstitial re-
gion. The LSDA technique includes an approximation
for both the exchange and correlation energies, and it
has been recently enhanced by the addition of elec-
tron density gradient terms to the exchange-correlation
energy. This has led to a generalized gradient approxi-
mation (GGA), as suggested by Perdew et al. [20.8], and
we used this improved method rather than the LSDA
approach.
20.2.2 Gibbs Energies of Solution Phases
Derived by the First-principles
Calculations
The formation enthalpies of some binary ordered struc-
tures derived by the first-principles calculations are
compared with experimental data in Table 20.2 [20.9].
In this comparison, alloy systems where the thermo-
dynamic analysis had already been completed, are
Part E 20.2