Горбунова В.Н., Савельева-Васильева Е.А., Красильников В.В. Молекулярная неврология. Заболевания нервно-мышечной системы
Подождите немного. Документ загружается.

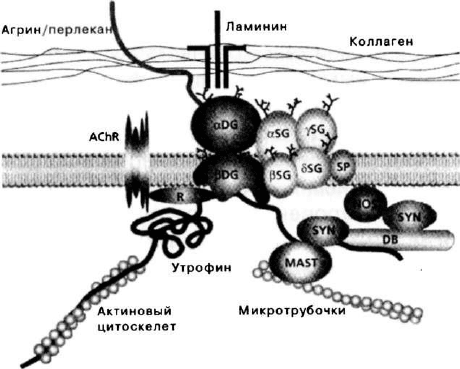
Индукция образования больших кластеров происходит с уча-
стием а-дистрогликана, по-видимому, посредством его взаи-
модействия с ламинином (Chamberlain, 1999). Утрофин мо-
жет играть роль стабилизатора подобных кластеров для уста-
новления точечных контактов между рецепторными агрега-
тами и цитоскелетом. Трансгенное разрушение рапсинового
гена у мышей сопровождается образованием аберрантных
нейромышечных соединений в результате потери способно-
сти к кластерированию ацетилхолиновых рецепторов, что и
приводит к ранней гибели животных (Gautam et al., 1995).
Таким образом, рапсин обеспечивает связь между ацетил-
холиновыми рецепторами и агрин-связанным утрофин-ассо-
циированным гликопротеиновым комплексом (Apel et al.,
1995). Еще одним отличием утрофин-ассоциированного ком-
плекса белков является его ассоциация с микротрубочками,
осуществляемая посредством взаимодействия цистеин-бо-
гатого домена утрофина со специфической серин/треонин-
киназой (MAST).
Рис. 9. Структура утрофин-ассоциированного комплекса
белков в нейромышечных соединениях

Интересно подчеркнуть, что з мышцах пациентов с
миодистрофией Дюшенна утрофин присутствует в сарко-
лемме, причем в положениях, характерных для дистрофи-
на. Это связано с посттрансляционными изменениями, а
не с увеличением скорости его транскрипции. Подобные
нарушения в регуляции распределения утрофина наблю-
даются при дерматомиозитах и полрмиозитах. По-видимо-
му, перераспределение утрофина возникает при регене-
рации мышцы и может служить компенсаторным механиз-
мом, частично защищающим мышечное волокно от даль-
нейших раундов дегенерации и регенерации.
9. Линии мышей, моделирующие миодистоофию
Дюшенна/Беккера
Успешному анализу особенностей организации гена
дистрофина и изучению молекулярных основ патогенеза
миодистрофии Дюшенна способствовали параллельные ис-
следования, выполненные на модельной линии мышей —
mdx. Эта модель была получена в результате отбора мута-
ции, спонтанно возникшей в одной из линий мышей дикого
типа (Bulfield et al., 1984). В мышцах и в мозге у мышей
этой линии резко снижено количестЕО Dmd-мРНК и не об-
наруживаются иммунологические формы дистрофина.
Идентифицирована нонсенс-мутация в экзоне 23 гена Dmd
у животных линии mdx, в результате <оторой транслирует-
ся лишь 27% дистрофинового полипептида. У mdx мышей,
так же как у больных с миодистрофией Дюшенна, значи-
тельно повышен уровень сывороточной креатинкиназы.
Несмотря на это, заметных фенотипинеских отклонений от
нормы у мутантных мышей не наблодается. Однако при
гистологическом исследовании скелетных мышц конечно-
стей и диафрагмы животных обнаруживаются зоны экстен-
сивной дегенерации и регенерации с многочисленными
патологическими признаками мышечной дистрофии, вклю-
чая некрозы, вариации по величине мышечных волокон,
центральную, а не периферическую локализацию ядер в
72
регенерирующих волокнах. У модельных mdx мышей, так
же как и у пациентов с миодистрофией Дюшенна/Беккера,
оказывается разрушен ассоциированный с дистрофином
гликопротеиновый комплекс.
Дополнительно путем индуцированного мутагенеза и
селекции были получены еще четыре линии mdx-мышей, в
которых проведена молекулярная идентификация мутантных
аллелей в гене Dmd (Сох et al., 1993b; lm et al., 1996). Фено-
тип мышей всех пяти mdx-линий по характеру мышечной па-
тологии очень сходен. Из-за присутствия различных точко-
вых мутаций в гене Dmd животных этих линий ни в их мыш-
цах, ни в мозге не экспрессируется полноразмерный дистро-
фии. Две из этих мутаций нонсенс типа, то есть создают преж-
девременный стоп-кодон, а три нарушают процесс сплайсин-
га первичного РНК-транскрипта. В результате различного
расположения мутаций относительно внутренних промоторов
гена Dmd исследуемые мутантные линии различаются по
набору экспрессирующихся укороченных изоформ дистрофи-
на. Таким образом, эти генетические линии представляют
собой идеальную систему для анализа функций множествен-
ных изоформ дистрофина.
У мутантных мышей, не экспрессирующих основной
продукт гена DMD в немышечных тканях — апо-дистрофин-1,
уровень дистрофин-ассоциированных белков в мозге значи-
тельно снижен, что указывает на важную роль этой укоро-
ченной изоформы дистрофина для формирования и/или ста-
билизации DAP-комплекса в центральной нервной системе
(Greenberg et al., 1996).
На базе mdx-мышей были сконструированы трансген-
ные линии животных, в которых при отсутствии дистрофи-
на наблюдается эктопическая экспрессия Dp71 (Сох et al.,
1994; Greenberg et al., 1994). Оказалось, что, в отличие от
mdx-мышей, у трансгенных животных апо-дистрофин-1 при-
сутствует в сарколемме скелетных мышц. Кроме того, при
экспрессии Dp71 содержание дистрофин-ассоциированных
белков — 59DAP, 50DAG и 43DAG — поднимается до нор-
73
мальных уровней и апо-дистрофин-1 оказывается спосо-
бен таким же образом, как и дистрофин, взаимодейство-
вать с этими белками, реконструируя дистрофин-гликопро-
теиновый комплекс. Несмотря на это, признаки мышечной
дистрофии, характерные для mdx-мышей — некрозы, ва-
рьирующая величина миофибрилл и центральное располо-
жение ядер в регенерирующих волокнах, а также высокий
уровень сывороточной креатинкиназы, не только сохраня-
ются при трансгенозе Dp71, но даже в некоторой степени
усиливаются. Таким образом, само по себе восстановле-
ние дистрофин-гликопротеинового комплекса недостаточ-
но для предотвращения мышечной дистрофии. Для сохра-
нения целостности мышечной мембраны, по-видимому,
необходимо взаимодействие N-концевого участка дистро-
фина с актиновыми филаментами цитоскелета миофиб-
рилл.
Отсутствие клинических проявлений миодистрофии у
mdx мышей частично может быть объяснено компенсатор-
ным действием утрофина. В частности, с этим, по-видимо-
му, связан тот парадоксальный факт, что у mdx-мышей, в
отличие от больных миодистрофией Дюшенна, не наблю-
дается сердечной недостаточности. Действительно, в сер-
дечной мышце мышей в норме уровень утрофина оказал-
ся в четыре раза выше, чем у человека. У дистрофин-де-
фицитных мышей, так же как и у пациентов с миодистро-
фией Дюшенна/Беккера, содержание утрофина в сарколем-
ме мышц значительно повышено, причем это связано с
посттрансляционными изменениями, а не с увеличением
скорости его транскрипции.
Для изучения роли комплементарного действия дис-
трофина и утрофина в патогенезе миодистрофии Дюшенна
была сконструирована серия трансгенных линий мышей с
различными сочетаниями дефектов в генах Dmd и Utrn
(Deconinck et al., 1997; Grady etal., 1997; Gilbert etal., 1998;
Rafael et al., 1998) (табл. 9). Оказалось, что аденовирусное
введение трансгена, специфически экспрессирующего в
74
скелетных мышцах укороченную форму утрофина (Tg Utm),
приводит к коррекции гистологических аномалий в линиях
mdx (Tinsley et al., 1996; Deconinck et al., 1997c). Укорочен-
ный трансген кодирует 200-кд белок, содержащий функци-
онально значимые амино- и карбокситерминальные доме-
ны утрофина, но не имеющий большей части его стержне-
вого спектрин-подобного района. Трансген работает под
контролем а-актининового промотора, обеспечивающего
специфическую экспрессию в скелетных, но не в сердеч-
ной мышце. В диафрагме и в конечностях мышей линии
(Tg Utm; mdx) экзогенный утрофин располагается в сарко-
лемме мышц. У трансгенных животных, в отличие от mdx-
мышей, значительно сокращена площадь некротических зон
и миофибрилл с варьирующей величиной и центральным
расположением ядер в регенерирующих волокнах скелет-
ных мышц. Кроме того, у них понижено содержание сыво-
роточной креатинкиназы и улучшены механические свой-
ства мышц, что определяется по многим показателям,
включая уровень спонтанной активности животных. Пол-
ностью восстанавливается нормальный внутриклеточный
гомеостаз кальция, нарушенный у mdx-мышей и у пациен-
тов с миодистрофией Дюшенна. При трансгенозе укорочен-
ных форм утрофина, по-видимому, происходит восстанов-
ление дистрофин-ассоциированного комплекса белков, так
как при иммуногистохимическом анализе биоптатов мышц
таких животных с использованием анти-ос-саркогликановых
антител появляется специфическое окрашивание в сарко-
лемме.
Инактивация гена Utrn у мышей не приводит к каким-
либо заметным фенотипическим аномалиям (Deconinck et al.,
1997b). Животные с (1Лгп-/-)-генотипом развиваются нормаль-
но и имеют незначительные дефекты, обусловленные неболь-
шими постсинаптическими аномалиями в нейромышечных
соединениях. При скрещивании мышей из линий mdx и
Utrn-/- удалось получить животных, у которых одновременно
Дефектными оказались оба гена — Dmd и Utrn (Grady et al.,
75
1997a; 1997b). В отличие от мышей родительских линий, осо-
би линии (Utrn-Л; mdx), называемой также Dko, представля-
ют из себя истинную фенокопию миодистрофии Дюшенна. У
таких животных наблюдается тяжелая мышечная дистрофия
с контрактурами суставов, кардиомиопатией, заметной за-
держкой роста, кифозом. Погибают они на сроке от 6 до 20 не-
дель постнатального развития. У мышей линии Dko при ульт-
развуковом исследовании наблюдаются структурные анома-
лии в нейромышечных и мышечно-сухожильных соедине-
ниях. При этом в нейромышечных соединениях белки дис-
трофин-ассоциированного комплекса — р-дистрогликан и
р2-синтрофин, а также белки ацетилхолино-рецепторных
(AchR) комплексов, в том числе рапсин, имеют нормаль-
ную локализацию. Таким образом, у двойных мутантов на-
блюдается тяжелая прогрессирующая мышечная дистрофия
при сохранении качественно нормального развития синап-
тической системы.
Удивительным оказалось то, что дистрофин/утрофин-
дефицитные животные с введенным трансгеном, экспрес-
сирующим в скелетных мышцах укороченную форму утро-
фина, не имеют аномалий, характерных для мышей линии
(Utrn-/-; mdx) (Rafael et al., 1998). Они нормально развива-
ются и даже спустя год не проявляют каких-либо клиниче-
ских симптомов миодистрофии, несмотря на то что в сер-
дечной мышце таких животных дистрофин и утрофин пол-
ностью отсутствуют. Показано также, что, в отличие от дис-
трофин/утрофин-дефицитных животных, у мышей линии (Тд
Utrn; Utrn-/-; mdx;) появляется окрашивание на а-сарко-
гликан в сарколемме мышц и восстанавливается экспрес-
сия некоторых белков, необходимых для нормального раз-
вития мышц. Очевидно, что присутствие укороченной фор-
мы утрофина в сарколемме мышц мутантов, не имеющих
нормального дистрофина и утрофина, способно восстано-
вить дистрофин-ассоциированный комплекс белков, нор-
мальное развитие и функционирование дефектных мышц.
76

Таблица 9
Линии
мышей
Характер
дефекта
Экспрессия
дистрофина
Экспрессия
утрофина
Фенотип
mdx
точковые
мутации
в Dmd
укороченная
форма
норма
физически
нормальны,
гистологические
аномалии мышц
TgUFrn;
mdx
точковые
мутации
в Dmd +
трансген
Utrn
укороченная
форма
норма +
укороченная
форма
в скелетных
мышца
физически
нормальны,
коррекция
гистологических
аномалий мышц
Шгп -/-
«нокаут»
Utrn
норма
отсутствие
экспрессии
физически
нормальны,
небольшие
постсинапти-
ческие
аномалии в
нейромышечных
соединениях
Utrn -/-;
mdx
точковые
мутации
в Dmd +
«нокаут»
Utrn
укороченная
форма
отсутствие
экспрессии
тяжелая форма
миодистрофии с
контрактурами
суставов,
задержкой роста,
кифозом и преж-
девременной
смертью
Tg utrn;
Utrn(-/-);
mdx
точковые
мутации
в Dmd +
«нокаут»
Utrn +
трансген
Utrn
укороченная
форма
отсутствие
экспрессии +
укороченная
форма в
скелетных
мышца
физически
нормальны,
значительная
коррекция
миодистрофии
и сопут твующих
симптомов,
небольшие
нейромышечные
дефекты
Характеристика мышей,
дефектных по дистрофиновому (Dmd)
и/или утрофиновому (Utrn) генам
Мы уже подчеркивали критическую роль дистроглика-
нов в поддержании всего дистрофин-ассоциированного ком-
плекса белков. Трансгенные мыши, гомозиготные по нуле-
вым аллелям дистогликанового гена (Dag1neo2), погибают в
раннем эмбриогенезе в результате грубых пороков разви-
тия, возникающих вследствие разрушения Рейхеротовской
мембраны. У таких эмбрионов нарушена локализация двух
критических структурных элементов этой оболочки — лами-
нина и коллагена IV.
Недавно были получены химерные мыши с недостаточ-
ностью дистрогликанов, путем инъекции в бластоцисты эмб-
риональных стволовых клеток (ЭСК), в которых предваритель-
но в условиях культивирования были направленно разруше-
ны оба аллеля гена Dag1 (Cote et al., 1999). У таких мышей
развивается прогрессирующая патология мышц, имеющая
большое сходство с тяжелой миодистрофией человека. У них
наблюдается выраженный кифосколиоз и деформация конеч-
ностей при нормальном развитии костной системы. В боль-
шинстве мышечных волокон химерных мышей дистрофин-ас-
социированный комплекс белков полностью отсутствует и не
наблюдается иммуноокрашивания на а-дистрогликан, дистро-
фин или а-саркогликан. Удивительным оказалось то, что при
этом сохраняется ультраструктура базальных мембран мышц
с правильной локализацией в них ламинина. Фенотип химер-
ных мышей во многих отношениях напоминает фенотип двой-
ных мутантов по дистрофину и утрофину. Однако, в отличие
эт мышей линии (Utrn-/-; mdx), у химерных животных оказа-
1ись разрушены многие нейромышечные соединения. Это
эще раз доказывает ведущую роль в развитии синаптичес-
:ой системы р-дистрогликана и, возможно, р2-синтрофина,
1рисутствующих у двойных мутантов по дистрофину и утро-
Ьину и отсутствующих у химерных животных. По-видимому,
истрогликаны совершенно необходимы для выживания мы-
ючных волокон, дифференцировки нейромышечных соеди-
ений и синаптогенеза, но не являются обязательными для
юрмирования базальных мембран мышц.
78
С использованием методов трансгенного моделирова-
ния исследовали роль нейрональной синтетазы окиси азота
(nNOS) в патогенезе миодистрофии Дюшенна. При отсутст-
вии дистрофина у больных, также как и у mdx-мышей, nNOS,
соединенная в норме с сарколеммой через комплекс дис-
трофин-гликопротеиновых белков, занимает неправильное
положение внутри мышечного волокна, где она продолжает
продуцировать окись азота. Было высказано предположение,
что одним из элементов сложного патогенетического пути при
миодистрофии Дюшенна является токсичность свободных
радикалов, обусловленная неправильной цитозольной лока-
лизацией nNOS. Однако это предположение не нашло под-
тверждения, так как у трансгенных mdx-мышей с инактиви-
рованным геном nNOS (nNOS-дистрофин нулевые мутанты)
сохраняются основные патологические характеристики mdx
мышей (Crosbie et al., 1998).
В заключение хотелось бы упоминуть об очень перспек-
тивных экспериментах по медикаментозной коррекции ано-
малий мышц в линии мышей mdx (Barton-Davis et al., 1999).
Мы уже упоминали о том, что причиной развития дистрофи-
ческого процесса у этих животных является нонсенс-мута-
ция в гене Dmd. Оказалось, что в присутствии антибиотика
гентамицина может происходить ошибочное проскакивание
стоп-кодона при трансляции белков. По-видимому, введение
определенных доз этого препарата в развивающиеся мыш-
цы мутантных животных может в каком-то проценте случаев
приводить к проскакиванию нонсенс-мутации в гене Dmd и
продолжению синтеза белка с образованием функциональ-
ного дистрофина. Напомним, что нонсенс-мутации в гене DMD
являются частой причиной развития миодистрофии Дюшен-
на и возможность гентамициновой терапии подобных случа-
ев заболевания представляется очень заманчивой. По-ви-
димому, в самое ближайшее время будут начаты клиничес-
кие испытания возможности такого лечения не только в от-
ношении миодистрофии Дюшенна, но и, возможно, других мо-
ногенных заболеваний, обусловленных ошибочным образо-
79

зованием стоп-кодонов и преждевременным прекращением
трансляции соответствующих белков.
10. Генотерапия миодистрофии Дюшенна/Беккера
В последние годы большое внимание уделяется разра-
ботке методов генотерапии миодистрофии Дюшенна. Эти ис-
следования пока не достигли стадии клинических испытаний.
Они проводятся на мутантных культурах клеток и модельных
линиях экспериментальных животных, в первую очередь на
линиях мышей mdx, а также на одной из линий охотничьих
собак (золотых ретриверов), у которых также обнаружена
мутация в гене дистрофина. При этом были опробованы раз-
личные способы введения генетического материала, вклю-
чая перенос миобластов, использование ретровирусных и
аденовирусных векторов, а также прямую инъекцию плаз-
мидной ДНК в мышцы.
Успешный перенос гена дистрофина в составе ретро-
вирусного вектора выполнен на культуре клеток млекопита-
ющих (Dickson et al., 1991). Наиболее перспективными для
переноса 14кб кДНК дистрофина, по-видимому, являются
аденовирусные мини-хромосомы, сочетающие в себе элемен-
ты плазмидной ДНК, аденовирусные сигналы начала репли-
кации и сигналы упаковки, а также содержащие р-галактози-
дазный ген-репортер и регуляторные элементы полноразмер-
ного гена дистрофина (Kumar-Singh, Chamberlain, 1996). В
серии экспериментов на mdx-мышах доказана принципиаль-
ная возможность генокоррекции миодистрофии Дюшенна
(Acsadi et al., 1991). Появление дистрофина человека в сар-
колемме мышечных волокон mdx-мышей наблюдали после
введения ретровирусных или аденовирусных генно-инженер-
ных конструкций, содержащих полноразмерную кДНК гена
дистрофина (14 кб) или его делетированную, но функциональ-
но активную форму, так называемый мини-ген размером
6.3 кб (Wells etal., 1992; Сох et al., 1993а; Acsadi et al., 1991;
1995). Мини-ген был сконструирован на базе кДНК пациента
с очень мягким течением миодистрофии Беккера, вызван-
80