Горбунова В.Н., Савельева-Васильева Е.А., Красильников В.В. Молекулярная неврология. Заболевания нервно-мышечной системы
Подождите немного. Документ загружается.


лин-3 или кавеолин М — специфичная для мышц форма се-
мейства белков, являющихся главными компонентами каве-
ол — присутствующих в большинстве типов клеток субком-
партментов плазматических мембран. Кавеолы являются
динамическими структурами, участвующими в выполнении
целого ряда функций, включая эндоцитоз и сигнальную транс-
дукцию. Предполагается, что кавеолины (VIP-21) играют
структурную роль в образовании неклатриновых везикул, в
том числе в образовании кавеоловых мембран, организуя и
концентрируя специфические кавеолин-взаимодействующие
липиды и белки на кавеоловых микродоменах.
Подобно дистрофину, кавеолин-3 локализован в сарко-
лемме мышечных волокон. При субклеточном фракциониро-
вании кавеолин-3 осаждается вместе с белками дистрофин-
ассоциированного комплекса и иммунопреципитируется дис-
трофиновыми антителами, что указывает на то, что кавео-
лин-3 и дистрофии являются частью дискретного комплекса.
При анализе гена CAV3 у 82 пациентов с аутосомно-доми-
нантной формой прогрессирующей конечностно-поясной
миодистрофей были найдены две миссенс-мутации — G55S
и C71W (McNally et al., 1998).
Предполагается, что мутации в гене CAV3 влияют на
процесс олигомеризации кавеолина-3 и на взаимодействие
с некоторыми кавеолин-ассоциированными сигнальными
молекулами, такими как гетеротримерные G-белки, H-Ras,
Src-семейство тирозинкиназ, эпидермальный фактор рос-
та R, протеинкиназа С и синтетаза окиси азота (eNOS). В
результате нарушается образование кавеол в плазматичес-
кой мембране мышечных клеток, что и ведет к развитию дан-
ной формы конечностно-поясной миодистрофии.
3. Конечностно-поясная миодистрофия. аутосомно-
реиессивная. форма LGMD2A
При генетическом анализе группы семей французско-
го происхождения с мягкой формой конечностно-поясной
миодистрофии было обнаружено сцепление локуса LGMD2A
с маркером D15S25, локализованным в проксимальной час-
91
ти длинного плеча хромосомы 15 (Beckmann et al., 1991).
Последующая идентифицикация фланкирующих маркеров,
расположенных на расстоянии около 7 сМ друг от друга, поз-
волила изолировать YAC-клоны, содержащие LGMD2A-reH, и
использовать их для уточнения цитогенетической локализа-
ции гена в области 15q15.1-q21.1 методом флюоресцентной
гибридизации in situ (Fougerousse et al., 1994). В дальнейшем
в этой области был идентифицирован ген CAPN3 (caleain-3),
ответственный за развитие данной формы конечностно-по-
ясной миодистрофии (Richard et al., 1995). CAPN3 кодирует
большую субъединицу кальций-активируемой нейтральной
специфической мышечной протеазы — кальпаина-3. Коди-
рующая часть гена CAPN3 разделена на 24 экзона, занима-
ющих более 40 кб геномной ДНК. В гене идентифицировано
4 микросателлитных повтора и 14 Alu-последовательностей.
CAPN3 специфически экспрессируется в скелетных мышцах
с образованием мРНК-транскрипта размером 3.4-3.6 кб.
Обнаружено более двух десятков мутаций в гене CAPN3
у больных с формой 2А миодистрофии плечевого и тазового
пояса. Около половины из них приводят к преждевременной
терминации трансляции. Миссенс-мутации затрагивают кон-
сервативные остатки или аминокислоты, расположенные в
функционально значимых участках доменов. Необычным яв-
ляется то, что шесть разных мутаций найдено у пациентов из
одной маленькой изолированной инбредной популяции жи-
телей острова Реюньона (LaReunion). Данные генеалогичес-
кого анализа, свидетельствующие о присутствии в данном
случае «эффекта основателя», находятся в противоречии с
результатами молекулярного анализа. Это противоречие по-
лучило название парадокса Реюньона. Для его объяснения
предложена дигенная модель наследования формы 2А конеч-
ностно-поясной миодистрофии, не получившая пока экспе-
риментальных подтверждений.
Кальпаины относятся к семейству внутриклеточных
нелизосомных цистеиновых протеаз. Они включают два по-
всеместно экспрессирующихся белка — типа 1 и 2, два спе-
92

цифических желудочных белка и мышечную форму типа 3.
Кальпаины являются гетеродимерами, состоящими из боль-
шой субъединицы, кодируемой соответственно генами
CAPN1, CAPN2 и CAPN3, соединенной с маленькой субъеди-
ницей, общей для всего семейства. Большие субъединицы раз-
делены на 4 домена, один из которых (II) имеет сходство с
цистеин-протеазами, содержащими гистидиновые, цистеино-
вые и аспарагиновые остатки в активных сайтах. Домен IV
имеет кальций-связывающие сайты. Кроме того, в кальпаи-
не-3 присутствуют три уникальных района, два из которых
содержат сигнал ядерной транслокации. Обнаружение энзи-
матического, а не структурного дефекта при миодистрофии
является неожиданной находкой. Наиболее простым объяс-
нением может быть то, что данная мышечная протеаза уча-
ствует в деградации или дестабилизации структурных компо-
нентов цитоскелета, экстраклеточного матрикса или дистро-
финового комплекса. Более вероятной кажется возможность
участия этого белка в слиянии миобластов. Ядерная локали-
зация кальпаина-3 не исключает его роли в контроле генной
экспрессии. Однако все эти гипотезы требуют эксперимен-
тальной проверки. Независимо от реальных механизмов па-
тогенного действия мутаций в гене CAPN3, обнаружение де-
фекта в энзиматическом белке дает основание для оптимиз-
ма в плане разработки фармакотерапии для данной формы
конечностно-поясной миодистрофии.
4. Конечностно-поясная миодистрофия. аутосомно-
реиессивная. форма LGMD2B
В ряде семей с клинически сходной конечностно-пояс-
ной миодистрофией обнаружено сцепление мутантного гена
не с маркерами хромосомы 15, а с короткими полиморфны-
ми тандемными повторами, локализованными в области 2р13
короткого плеча хромосомы 2 — D2S134 и D2S136. Это ука-
зывает на существование другого гена — LGMD2B, мутации
в котором могут приводить к мягкой аутосомно-рецессивной
форме заболевания типа 2В (Bashir et al., 1994). В этой же
области генома локализован ген MM (Miyoshi myopathy), от-
93
ветственный за дистальную форму миопатии с аутосомно-
рецессивным типом наследования, известную как миопатия
Миоши. Болезнь дебютирует в ювенильном или молодом воз-
расте в виде слабости икроножных мышц.
Оказалось, что конечностно-поясная миодистрофия
типа 2В и миопатия Миоши являются аллельными заболева-
ниями, обусловленными мутациями в гене DYSF (dystrophy-
associated fer-1 -like) (Bashir et al., 1998). Продукт этого гена —
дисферлин — имеет высокий процент гомологии с белком
fer-1 Caenorhabditis elegans, дефектным у мутантов с нару-
шенным сперматогенезом. Дисферлин — цитоплазматичес-
кий белок, состоящий из 2080 аминокислот, имеет большой
заряженный гидрофильный район и один мембран-связыва-
ющий район на С-конце, обеспечивающий возможность его
ассоциации с саркоплазматическим ретикулумом (Liu et al.,
1998). В цитоплазматическом домене дисферлина идентифи-
цированы последовательности, характерные для белков, вза-
имодействующих с ядерной мембраной. Кроме того, цитоплаз-
матический компонент этого белка содержит 4 мотива, гомо-
логичные С2 доменам — внутриклеточным белковым моду-
лям, состоящим из 80—130 аминокислотных остатков и уча-
ствующим, по-видимому, в связывании кальция и фосфоли-
пидов. Обнаружено 9 мутаций в гене DYSF у пациентов с
дистальными и проксимальными формами конечностно-по-
ясной миодистрофии. 5 из этих мутаций приводят к прежде-
временной терминации трансляции. Имеется выраженный
фенотипический полиморфизм среди больных, несущих оди-
наковые мутации. Так, описаны семьи, у одних членов кото-
рой болезнь протекает по типу миопатии Миоши, тогда как у
других — в виде конечностно-поясной миодистрофии 2В.
Недавно было показано, что описанная более 30 лет
тому назад генетическая линия мышей SJL является естест-
венной моделью обсуждаемой формы конечностно-поясной
миодистрофии (Bittner et al., 1999). Мыши этой линии облада-
ют повышенной восприимчивостью к индуцируемым аутоим-
мунным болезням, таким как экспериментальные аутоиммун-
94

ные энцефалиты (ЕАЕ) и воспалительные болезни мышц. У
мутантных животных часто развиваются спонтанные воспа-
лительные миопатии, при этом в мышцах повышена способ-
ность к регенерации. На линии SJL на протяжении многих
лет проводился широкий объем экспериментальных иссле-
дований, касающихся различных аутоиммунных болезней
человека, изучались процессы регенерации и транспланта-
ции мышц.
С использованием набора из 40 микросателлитных мар-
керов мышиный ген, ответственный за фенотип мутантных
животных, был картирован в 6 хромосоме в области, синтен-
ной району локализации гена DYSF человека. Оказалось, что
у мутантных животных экспрессия дисферлина в мышцах
составляет не более15% по сравнению с нормой. Это послу-
жило основанием для изоляции и клонирования кДНК гомо-
логичного гена мыши — Dysf. При изучении кодирующей об-
ласти этого гена у мутантных животных была найдена гомо-
зиготная делеция 171 нуклеотида, сопровождающаяся отсут-
ствием протяженного участка четвертого С2 домена в соот-
ветствующем белке и нарушением протеолитической стабиль-
ности дисферлина. Таким образом, обнаружение в линии SJL
мутации, определяющей развитие прогрессирующей дегене-
ративной болезни мышц, требует пересмотра эксперимен-
тальных результатов, полученных на этой генетической ли-
нии животных.
5. Конечностно-поясная миодистрофия. аутосомно-
рецессивная. формы LGMD2C-2F (саркогликанопатии)
Гены, ответственные за более тяжелые формы аутосом-
но-рецессивной конечностно-поясной миодистрофии
LGMD2C — LGMD2F, кодируют белки саркогликанового дис-
трофин-ассоциированного субкомплекса: у-, а-, р- и 8-сар-
когликаны соответственно (Roberds et al., 1994; Lim et al., 1995;
Bonnemann et al., 1995; Noguchi et al., 1995; Nigro et al., 1996b).
Все четыре белка имеют ряд общих характеристик. Они N-
гликозилированы, содержат короткий внутриклеточный до-
95
мен, один трансмембранный и большой внеклеточный доме-
ны с С-терминальным кластером цистеиновых остатков. В
качестве примера на рис. 12 и 13 представлена структура (5-
и у-саркогликана соответственно с указанием локализации
мутаций, идентифицированных у больных с тяжелыми фор-
мами аутосомно-рецессивной конечностно-поясной миоди-
строфии. У всех пациентов с мутациями в любом из сарког-
ликановых генов отсутствует а-саркогликан. Это может быть
первичным дефектом при форме 2D или вторичным — при
формах 2С, 2Е и 2F. При этом наблюдается корреляция меж-
ду клиническим фенотипом и степенью сохранности сарког-
ликанового комплекса, определяемой главным образом ти-
пом мутационного повреждения. Независимо от формыза-
болевания у пациентов с тяжелым течением этот комплекс,
как правило, полностью разрушен. Ген а-саркогликана (ад-
халина) — ocSG (обозначаемый также как ADL или SGA или
DAG2), картирован в коротком плече хромосомы 17 в облас-
ти 17р21 (McNally et al., 1994). Мутации в этом гене приводят
к первичной адхалинопатии, варьирующей по степени тяже-
сти аутосомно-рецессивной мышечной дистрофии — аллель-
ному варианту конечностно-поясной миодистрофии LGMD2D
(Passos-Bueno et al., 1995).
В описанной ранее тунисской семье, так же как и в ряде
других случаев, обнаружено сцепление заболевания с мар-
керами, локализованными в области 13q12 (Ben Othmane et
al., 1992; Sewry et al., 1994). В этой области картирован ген у-
саркогликана, мутации в котором ответственны за развитие
тяжелой детской аутосомно-рецессивной мышечной дистро-
фии — аллельному варианту конечностно-поясной миодист-
рофии LGMD2C (Noguchi et al., 1995). У ряда пациентов с тя-
желым течением заболевания в тунисских, а также в четы-
рех бразильских семьях в гене ySG была обнаружена одно-
типная мутация — del521-T, сопровождающаяся сдвигом рам-
ки считывания — рис. 13 (Vainzof et al., 1996). У пациентов,
гомозиготных по этой мутации, у-саркогликан в биоптатах
мышц полностью отсутствует и в различной степени умень-
96
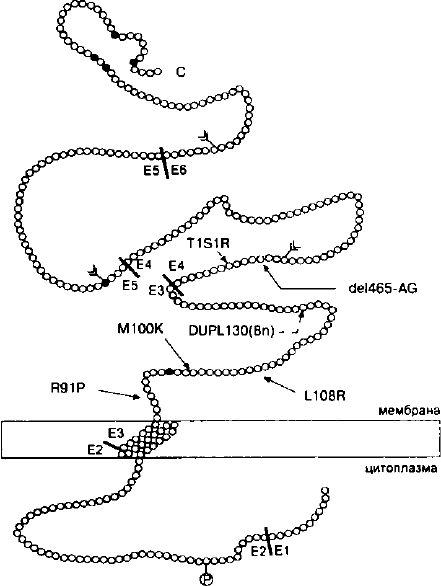
Рис. 12. Схематическое представление бета-саркогликана с
указанием мутаций, идентифицированных у пациентов с
формой 2Е конечностно-поясной миодистрофии
шено содержание а- и р-саркогликанов. Интересно отметить,
что у трех сибсов в одной из бразильских семей, в отличие от
всех других пациентов с данной мутацией, болезнь протека-
ла в очень мягкой форме и наблюдалась корреляции между
клиническим фенотипом и сохранением саркогликанового
комплекса при полном отсутствии у-саркогликана. При скри-
нинге мутаций в гене ySG у 50 пациентов из США и Италии с
7 Заказ № 170
97
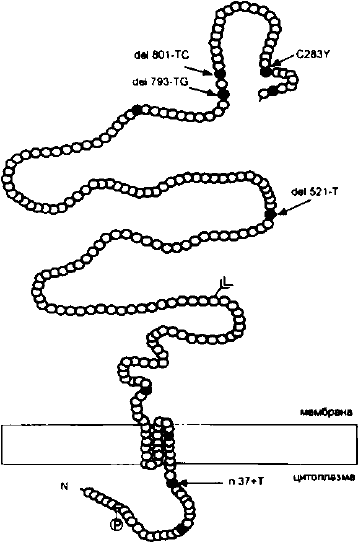
Рис. 13. Схематическое представление
гамма-саркогликана с указанием мутаций,
идентифицированных у пациентов с формой 2С
конечностно-поясной миодистрофии.
тяжелой формой аутосомно-рецессивной миодистрофии были
идентифицированы четыре гомозиготные мутации, сопровож-
дающиеся сдвигом рамки считывания (McNally et al., 1996).
Одна из них del521-T, другая — инсерция тимидина после
87 нуклеотида, и две оставшиеся мутации — микроделеции,
локализованные в З'-конце гена — del801-TG и del793-TG. У
пациентов с последними двумя мутациями полностью отсут-
ствует не только а- и у-, но и р-саркогликан. По-видимому,
98
карбокси-терминальный район у-саркогликана имеет важное
значение для сохранения стабильности всего саркогликано-
вого комплекса.
У пациентов с тяжелой дюшенно-подобной аутосомно-
рецессивной миодистрофией из больших неродственных цы-
ганских семей, проживающих в разных странах Западной
Европы, в гене ySG обнаруживается однотипная гомозигот-
ная миссенс-мутация — C283Y, затрагивающая консерватив-
ный цистеиновый остаток в карбокси-терминальном районе
экстраклеточного домена белка — рис. 13 (Piccolo etal., 1996).
Для этих больных характерны ранний дебют, повышенный
уровень сывороточной креатинкиназы, отсутствие а- и у-сар-
когликана в мышцах при нормальном уровне дистрофина. У
всех гомо- и гетерозиготных носителей данная мутация на-
ходится в абсолютном неравновесии по сцеплению с одним
из аллелей внутригенного высокополиморфного маркера
D13S232. Анализ полиморфизма по фланкирующим марке-
рам позволил определить гаплотип общего предка, у которо-
го мутация, по-видимому, возникла от 60 до 200 поколений
тому назад. Полученные данные согласуются с предположе-
нием о миграции предков цыган из Индии.
Картирован и клонирован ген р-саркогликана — PSG.
Он локализован в области 4q12, его кодирующая часть раз-
делена на 6 экзонов — рис. 12. Мутации в этом гене сопро-
вождаются разрушением саркогликанового субкомплекса и
реализуются в виде конечностно-поясной миодистрофии
формы 2Е (Lim et al., 1995) либо в виде аутосомно-рецессив-
ной дюшенно-подобной миодистрофии (Bonnemann et al.,
1995). Первые две мутации в этом гене, одна из которых при-
водит к сдвигу рамки считывания, а другая к образованию
преждевременного стоп-кодона, были идентифицированы в
спорадическом случае у девочки с дюшенно-подобной мио-
дистрофией (рис. 12). Одновременно у нескольких неродст-
венных пациентов из Южной Индианы с мягкой формой ко-
нечностно-поясной миодистрофии была обнаружена общая
миссенс-мутация — Т151R, находящаяся в гомозиготном со-
99
стоянии. При скрининге мутаций в гене (3SG у 15 неродствен-
ных пациентов бразильского происхождения с тяжелым те-
чением конечностно-поясной миодистрофии также были
обнаружены четыре мутации в двух семейных и в двух спо-
радических случаях (Bonnemann et al., 1996). Три из них
присутствовали у пациентов в гомозиготном состоянии и толь-
ко одна сопровождалась сдвигом рамки считывания. Примеча-
тельно, что все три мутации миссенс-типа были локализованы
в экзоне 3, кодирующем экстраклеточный домен — рис. 12.
При генетическом анализе негритянских семей бразиль-
ского происхождения с тяжелыми формами конечностно-пояс-
ной миодистрофии был обнаружен шестой ген, ответственный
за аутосомно-рецессивные формы заболевания (Passos-Bueno
et al., 1996b). В области 5q33-34 был идентифицирован ген —
8SG, кодирующий новый компонент саркогликанового субкомп-
лекса — 6-саркогликан (Nigro et al., 1996а). Все 8 пациентов из
4 неродственных бразильских семей оказались гомозиготны по
однотипной мутации —делеции одного нуклеотида в экзоне 7 —
del656C, приводящей к сдвигу рамки считывания (Nigro et al.,
1996b). При секвенировании полноразмерной кДНК пациентов
никаких других мутаций не было найдено. Таким образом, было
доказано, что именно этот ген ответственен за форму 2F ко-
нечностно-поясной миодистрофии.
Интересно подчеркнуть, что мутация в 6-саркогликано-
вом гене, активно экспрессирующемся в сердечной и ске-
летных мышцах, найдена в хорошо известной линии сирий-
ского хомячка — BI014.6, используемой в качестве генети-
ческой модели аутосомно-рецессивной кардиомиопатии. Для
животных этой линии характерны прогрессирующие некрозы
миокарда и сердечная недостаточность.
б) Мышечная дистрофия плечевого и тазового
пояса с буллезным эпидермолизом (MIM: 226670)
Необычная форма аутосомно-рецессивного буллезно-
го эпидермолиза в сочетании с мышечной дистрофией пле-
чевого и тазового пояса (MD-EBS — muscular dystrophy —
100