Горбунова В.Н., Савельева-Васильева Е.А., Красильников В.В. Молекулярная неврология. Заболевания нервно-мышечной системы
Подождите немного. Документ загружается.

Наиболее частый тип мутаций в гене STA у пациентов с
миодистрофией Эмери-Дрейфуса — небольшие внутриген-
ные структурные перестройки. Идентифицированы также и
протяженные делеций, включающие целиком ген эмерина
(Small et al., 1998). Все описанные к настоящему времени
мутации приводят либо к полному отсутствию, либо к обра-
зованию укороченного продукта STA-гена вследствие преж-
девременной терминации трансляции. Содержание эмерина
у женщин носительниц мутаций в гене STA часто значимо
отклоняется от 50%, что свидетельствует о разном уровне экс-
прессии нормальных и мутантных аллелей (Manilal et al., 1998).
Полное секвенирование насыщенного кодирующими
последовательностями 219-кб района Xq28 между локусами
цветовой слепоты (RCP/GCP) и глюкозо-6-фосфат-
дегидрогеназы (G6PD) (Chen et al., 1996) позволило расшиф-
ровать молекулярный механизм возникновения делеций все-
го гена STA, идентифицированной у одного из пациентов с
мышечной дистрофией Эмери-Дрейфуса (Small et al., 1997).
В непосредственной близости от STA со стороны центроме-
ры расположен 26-кб филаминовый ген (FLN1), разделенный
на 48 экзонов и кодирующий актин-связывающий белок 280.
Фланкируют этот 48-кб FLNI/STA-район два больших инвер-
тированных повтора размером 11.3 кб. Уровень гомологии
между этими повторами превосходит 99%. Оказалось, что
причиной возникновения делеций является ошибочное спа-
ривание между большими инвертированными повторами с
последующей двойной рекомбинацией между рядом ошибоч-
но спаренных повторов и внутренними последовательностя-
ми ДНК. Более частым следствием этих событий является
инверсия FLN1/STA области, присутствующая в гетерозигот-
ном состоянии у 33% женщин. Внутрихроматидная рекомби-
нация вследствие неправильного спаривания двух инверти-
рованных повторов служит причиной возникновения частых
инверсий, зарегистрированных также при тяжелых формах
гемофилии А и синдрома Хантера (Lakich et al., 1993;
Lagerstedt et al., 1997).
111

4. Структура и функции эмерина
Ген STA кодирует серин-богатый белок, состоящий из
254 аминокислот, для которого не найдено гомологов сре-
ди других белков с известными функциями. Так как это
новый белок, авторы предложили называть его эмерином.
Два района эмерина — 39 аминокислот на N-конце и 34 —
на С-конце, обнаруживают сходство (41% гомологии) с
белками неизвестной функции — тимопоиетинами, что, в
свою очередь, обусловливает и некоторые его структур-
ные особенности. Кроме того, обнаружено сходство по
аминокислотной последовательности эмерина с ядерным
ламина-ассоциированным белком — LAP2 (Furukawa et al.,
1995). В целом белок гидрофильный, но имеет на С-конце
гидрофобный домен достаточной протяженности, чтобы
служить в качестве мембранного якоря. В эмерине при-
сутствует большое количество сайтов фосфорилирования
и только один возможный сайт N-гликозилирования. Сиг-
нального пептида не обнаружено. Идентифицирована так-
же и RGD-последовательность (аргинил-глицил-аспартил —
мотив клеточной адгезии), важная для взаимодействий
между белками внеклеточного матрикса. По-видимому,
эмерин может быть мембранным белком с коротким отри-
цательно заряженным С-хвостом и большим N-терминаль-
ным цитозольным доменом. Подобные белки, способные
заякореваться хвостовой частью на мембранах, обнару-
жены в различных клеточных компартментах. В том числе
большое количество таких белков (синаптобревин, синтак-
сины) присутствует в органеллах секреторного пути, вовле-
ченного в транспорт везикул. При биохимическом фрак-
ционировании показано полное отсутствие эмерина в рас-
творимых фракциях. Это согласуется с предположением о
том, что эмерин является одним из членов семейства ядер-
ных мембранных ламина-ассоциированных белков (LAPs).
На рис. 14 схематически представлена организация эме-
рина и тимопоэтинов на ядерной мембране. Показано, что
эмерин ассоциирован с внутренней мембраной ядер и эта
112
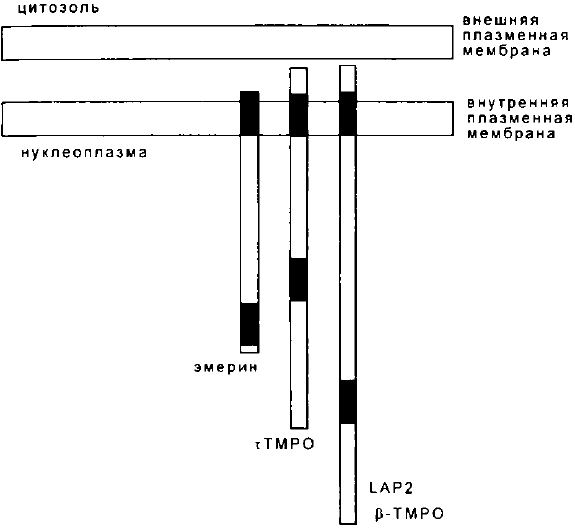
связь осуществляется посредством С-терминального гид-
рофобного домена (Cartegni et al., 1997).
Рис. 14. Организация эмерина и тимопоэтинов
на ядерной мембране
Большой фрагмент кДНК гена STA был введен в экс-
прессионную систему Е. coli, и продукт этой конструкции был
использован в качестве иммуногена для получения панели
из 12 моноклональных антител, реагирующих по крайней мере
с 4 различными антигенными детерминантами гидрофильно-
го района эмерина (Manilal et al., 1996). Во всех исследован-
ных тканях все варианты антител идентифицировали один и
тот же 34-кД белок, локализованный по краям ядер. В био-
8 Заказ № 170
113

птатах мышц пациентов с мышечной дистрофией Эмери-Дрей-
фуса этот белок не мог быть обнаружен ни методами имму-
ногистохимии, включая иммунофлюоресцентную микроско-
пию, ни с использованием метода Вестерн блот-гибриди-
зации. Таким образом, при наличии соответствующих ан-
тител иммунологические методы могут быть использова-
ны в качестве наиболее простых диагностических тестов
для данного заболевания. В сердце и в культивируемых кар-
диомиоцитах эмерин обнаруживается также в интеркаляр-
ных дисках.
5. Лопаточно-перонеальные синдромы (SP- синдромы)
Нередко пациентам с доброкачественным течением
миодистрофии ошибочно ставится диагноз лопаточно-перо-
неального синдрома или плече-перонеальной миопатии. Это
гетерогенная группа аутосомно-доминантных нейро-мышеч-
ных заболеваний, характеризующихся слабостью мышц пле-
чевого пояса и перонеальной мускулатуры. Клинически вы-
деляют две формы — нейрогенную (лопаточно-перонеальная
спинальная мышечная атрофия MIM: 181405, ген SPSMA) и
первично мышечную (лопаточно-перонеальная мышечная
дистрофия MIM: 181430, ген SPMD). Локус SPMD картиро-
ван в длинном плече хромосомы 12 (Wilhelmsen et al., 1996).
При генетическом анализе большой семьи с аутосомно-до-
минантной нейрогенной формой лопаточно-перонеального
синдрома ген SPSMA был картирован в области 12q24.1-
q24.31, примерно на 20 сМ ближе к теломере по сравнению с
локусом SPMD (Isozumi etal., 1996). Определен 19-сМ интер-
вал наиболее вероятной локализации гена между маркера-
ми D12S338 и D12S366, что является предпосылкой для иден-
тификации гена SPSMA методами позиционного клонирова-
ния. Антиципация по дебюту и тяжести течения заболевания,
наблюдаемая в исследуемой родословной, дает основания
предполагать возможное участие динамических мутаций в
генетическом контроле данного состояния. В непосредствен-
ной близости от области локализации гена SPSMA картиро-
114
ван ген SCA2, ответственный за одну из форм спиноцере-
беллярной атаксии, обусловленной экспансией внутриген-
ного CAG-повтора. В области поиска SPSMA локализова-
ны следующие гены: NOS1, продуктом которого является
нейротрансмиттер, участвующий в образовании окиси азо-
та и играющий важную роль в морфо- и синаптогенезе нерв-
ной системы; АТР5В, кодирующий АТФ-синтазу; АТР2А2/
АТР2В, кодирующий Са
2+
-зависимую АТФазу; MYL2, коди-
рующий легкую цепь 2 миозина. Эти гены рассматривают-
ся в качестве кандидатных для SPSMA.
Верификация диагноза миодистрофии Эмери-Дрейфу-
са в сомнительных случаях может быть достигнута только при
использовании иммунологических или молекулярно-генети-
ческих методов исследования. Однако необходимо иметь в
виду, что в ряде семей заболевание наследуется по аутосом-
но-рецессивному типу и не связано с дефектами эмерина.
Так, при полном секвенировании гена STA, выполненном у
пациентов из 17 семей с миодистрофией Эмери-Дрейфуса,
мутации не были обнаружены в трех семьях (Manilal et al.,
1998). При этом уровень эмерина у больных, принадлежа-
щих этим семьям, соответствовал норме.
б) Скапуло-илио-перонеальная атрофия с
кардиопатией (MIM: 181350)
Аутосомно-доминантная миодистрофия Эмери-Дрейфу-
са или скапуло-илио-перонеальная атрофия с кардиопатией
клинически не отличается от Х-сцепленной формы заболе-
вания. При генетическом анализе, выполненном с исполь-
зованием 280 высокоинформативных микросателлитных
маркеров в большой французской семье, насчитывающей
54 индивидуума, 17 из которых были больными, удалось кар-
тировать мутантный ген EDMD-AD (Emery-Dreifuss muscular
dystrophy — autosomal dominant) в области 1q11-q23 в 8-cM
интервале между маркерами D1S2346 и D1S2125 (Bonne et
al., 1999). Анализ гаплотипов по микросателлитным марке-
рам этого интервала, проведенный в четырех других семьях
115
с подобной формой миодистрофии Эмери-Дрейфуса, под-
твердил генетическую однородность исследуемой выбор-
ки больных.
Локализованный ранее в области 1q21.2-q21.3 ген
LMNA, кодирующий белки ядерной ламины — ламин А и С,
был исследован в качестве кандидатного гена для EDMD-AD.
С использованием методов прямого секвенирования двух
амплифицированных экзонов (1 и 7) и SSCP-анализа
остальных 10 экзонов были идентифицированы точечные
мутации в гене LMNA у пациентов всех пяти отобранных
семей, причем эти мутации отсутствовали у их здоровых
родственников. Одна из этих мутаций создавала
преждевременный стоп-кодон, три другие сопровождались
заменой консервативных аминокислот. При этом одна из
миссенс-мутаций — R527P — независимо встретилась в двух
неродственных семьях. Таким образом была доказана
идентичность генов LMNA и EDMD-AD.
Ламины А и С, образующиеся в результате альтерна-
тивного сплайсинга первичного РНК-транскрипта гена
LMNA, относятся к семейству промежуточных филамент,
присутствующих в клетках на стадии конечной дифферен-
цировки. Они формируют часть ядерной ламины — фиб-
розный слой на нуклеоплазменной стороне внутренней
ядерной мембраны, служащей каркасом для ядерной обо-
лочки. Ламины имеют стержневой домен, участвующий в
образованиии димеров,и карбокси-терминальный глобу-
лярный домен.Они взаимодействуют с хроматином и ин-
тегральными белками внутренней ядерной мембраны, в том
числе с эмерином. При иммуногистохимическом анализе
биоптатов сердечной мышцы было показано, что локализа-
ция ламина. А/С и эмерина у больных в исследуемых семьях
не изменена. По-видимому, идентифицированные мутации в
гене LMNA не приводят к серьезным нарушениям структуры
ядерной оболочки, но изменяют ее функции.
Ранее при генетическом анализе, выполненном в трех
семьях с аутосомно-доминантной конечностно-поясной ми-
116
одистрофией, ассоциированной с кардиопатией (LGMD1B),
мутантный ген был картирован в той же области 1q11-q22,
где расположен ген LMNA (van der Kooi et al., 1997). Несмот-
ря на значительные клинические отличия конечностно-пояс-
ной миодистрофии 1В от аутосомно-доминантной миодист-
рофии Эмери-Дрейфуса, выражающиеся в отсутствии кон-
трактур и гипертрофии икроножных мышц, а также в преиму-
щественной слабости проксимальных отделов конечностей,
не исключена аллельная природа этих заболеваний и воз-
можность участия в патогенезе LGMD1B каких-то иных спе-
цифических мутаций в гене LMNA. Возможно также, что еще
одна форма аутосомно-доминантной конечностно-поясной
миодистрофии- 1А-связана с мутациями в гене LMNB1, ко-
дирующим другой компонент ядерной ламины — ламин В1,
так как мутантный ген LGMD1А картируется в той же области
5q22.3-31.3 длинного плеча хромосомы 5, где расположен ген
LMNB1. Конечно, эти предположения требуют эксперимен-
тального подтверждения. Однако очевидно, что компоненты
ядерной мембраны вносят определенный вклад в патогенез
некоторых нейромышечных заболеваний.
1.6. Врожденные непрогрессирующие
миопатии
В самостоятельную клиническую группу традиционно
выделяют врожденные непрогрессирующие миопатии. Две из
них обусловлены дефектами белков базальной ламины. При
врожденных миопатиях дефектными, как правило, оказыва-
ются гены, экспрессирующиеся в раннем эмбриогенезе и
кодирующие белки, участвующие в процессах роста, диффе-
ренцировки и пролиферации миобластов. Примерно в поло-
вине случаев врожденных аутосомно-рецессивных мышеч-
ных дистрофий с ранним дебютом у больных обнаруживает-
ся недостаточность по а2-цепи ламинина 2 (мерозина), лока-
лизованного в базальной мембране поперечно-полосатых
мышц и связанного с цитоскелетом клетки через комплекс
дистрофин-ассоциированных белков. В некоторых случаях это
117
снижение является вторичным, как например при миодист-
рофии Фукуяма. Мутации в гене ламинина 2 приводят к раз-
витию врожденной мерозин-дефицитной миодистрофии, со-
четающейся с гипомиелинизацией белого вещества мозга.
Еще одна редкая форма непрогрессирующей врожденной
миопатии вызвана наследственной недостаточностью рецеп-
тора ламинина в мышцах — а7 интегрина (31D.
Мы уже упоминали о том, что патологические процес-
сы при некоторых врожденных миопатиях сопровождаются
образованием в клетках гистологически идентифицируемых
аномалий. Так, при немалиновой миопатии в мышечных клет-
ках пациентов присутствуют нитеобразные патологические
фибриллярные структуры, причиной развития которых явля-
ется латеральная экспансия Z-дисков. Эта экспансия при
различных формах немалиновой миопатии может возникать
вследствие нарушений в структуре интегральных белков тон-
ких филамент, таких как небулин и актин, а также других вза-
имодействующих с ними белков, в первую очередь тропоми-
озина В. Определенные гистологические аномалии в мышеч-
ных клетках характерны также для пациентов с болезнью
центрального стержня и с миотубулярной миопатией. В обо-
их случаях дефектными оказываются белки, участвующие в
контроле дифференцировки мышечных волокон. Различные
варианты еще одной врожденной миопатии Бетлема обуслов-
лены дефектной структурой микрофибриллярного коллагена
VI типа, оказывающего влияние как на процессы дифферен-
цировки миобластов, так и на структурную организацию вне-
клеточного матрикса.
а) Мышечная дистрофия врожденная мерозин-
дефицитная (MIM: 156225)
В «классическом» варианте мерозин-дефицитной мио-
патии клинические проявления ограничиваются только ске-
летными мышцами без каких-либо симптомов поражения цен-
тральной нервной системы, хотя при магнитно-резонансной
томографии выявляются изменения в белом веществе моз-
118
га, обусловленные гипомиелинизацией (Banker, 1994; Mercuri
et al., 1995). Болезнь проявляется в виде мышечной слабос-
ти, гипотонии, задержки моторного развития. Дети поздно на-
чинают садиться и ходить, при этом оказываются не способ-
ны передвигаться по неровной местности и на большие рас-
стояния без посторонней помощи. В сыворотке крови наблю-
дается повышенное содержание креатинкиназы. В биопта-
тах мышц больных отмечаются гистологические аномалии,
свидетельствующие об умеренном течении дистрофическо-
го процесса.
Ламинин-2 преимущественно локализован в базальной
мембране поперечно-полосатых мышц. Он состоит из тяже-
лой ос2 цепи и двух легких цепей (J1 и у1, кодируемых разны-
ми генами. Ламинин-2 экспрессируется достаточно поздно в
эмбриогенезе и на более ранних стадиях его функции, по-
видимому, выполняет ламинин-1. Ламинин-2 является есте-
ственным лигандом для а-дистрогликана. Зрелая а-цепь ла-
минина-2 состоит из 3088 аминокислотных остатков, сигналь-
ный пептид содержит 22 аминокислоты (Vuolteenaho et al.,
1994). При некоторых формах врожденных мышечных дис-
трофий содержание ламинина-2 в скелетных мышцах и в пе-
риферических нервах резко снижено. В некоторых случаях
это снижение является вторичным.
При исследовании 20 семей с врожденной аутосомно-
рецессивной миодистрофией, не относящейся к типу Фукуя-
ма, в 13 было обнаружено полное отсутствие ламинина-2
(Hillaire et al., 1994). Последующий генетический анализ, про-
веденный в этих мерозин-дефицитных семьях, с использо-
ванием метода картирования у пациентов областей генома,
гомозиготных по высокополиморфным микросателлитным
маркерам, показал, что мутантный ген, ответственный за
данную форму врожденной миодистрофии, локализован в той
же области 6q22, где расположен ген LAMA2 (laminin alpha
2). В дальнейшем локализация гена LAMA2 в области 6q22-
q23 была подтверждена многими методами, включая метод
флюоресцентной гибридизации in situ (Vuolteenaho etal., 1994).
119
LAMA2 — это очень крупный ген, занимающий 260 кб
геномной ДНК. Его кодирующая часть разделена на 64 экзо-
на, два из которых очень маленькие — 6 и 12 п.о. (Zhang et
al., 1996). Идентификация специфических мутаций в гене
LAMA2 у пациентов явилась окончательным доказательством
его причастности к развитию болезни (Helbling-Leclerc et al.,
1995; Nissenen et al., 1996; Allamand et al., 1997). Различные
мутации в гене LAMA2 могут приводить к разным клиничес-
ким формам заболевания от тяжелых, при которых ламинин-
2 полностью отсутствует, до промежуточных и мягких, при
которых наблюдается количественное снижение белка или
присутствуют его дефектные варианты. Так, у двух родствен-
ных пациентов с мягким течением врожденной мышечной
дистрофии обнаружена сплайсинговая мутация, сопровож-
дающаяся делецией 63 аминокислот в IV домене белка. Этот
домен ответственен за формирование глобулярной структу-
ры на коротком плече сс2-цепи ламинина-2, отсутствие кото-
рой, по-видимому, сопровождается разрушением ламининовой
сети в мышечных мембранах и нарушением связи белка с дис-
трофин-гликопротеиновым комплексом (Allamand et al., 1997).
При молекулярном обследовании родственников боль-
ных с мерозин-дефицитной миодистрофией выявляется до-
стоверное превышение гетерозиготных носителей мутаций в
гене LAMA2. Для объяснения наблюдаемого преимущества
гетерозигот привлекается гипотеза отбора, происходящего
либо на уровне гамет, либо среди ранних зародышей на до-
имплантационной стадии развития.
Мутация в гомологичном гене Lama2, кодирующем а2-
цепь ламинина мышей, описана у животных генетической
линии dy2j с мягкими проявлениями мышечной дистрофии
(Xu et al., 1994). Эта
ЛИНИЯ
является
одним из
аллельных
ва-
риантов классической линии мышей с врожденной мышеч-
ной дистрофией — dy, впервые описанной почти 40 лет на-
зад. Показано, что молекулярной основой наблюдаемого
фенотипа в линии dy2j является нарушение процесса сплай-
синга первичного РНК транскрипта Lama2 и трансляция по-
120