Горбунова В.Н., Савельева-Васильева Е.А., Красильников В.В. Молекулярная неврология. Заболевания нервно-мышечной системы
Подождите немного. Документ загружается.

ретракции отсутствуют. Течение заболевания доброкачест-
венное, отмечены даже улучшения в виде нарастания мы-
шечной силы в юношеском возрасте. Витальный прогноз бла-
гоприятный. В ряде семей болезнь центрального стержня
сочетается со злокачественной гипертермией — аутосомно-
доминантно наследуемой повышенной чувствительностью к
анестезии, обусловленной, по-видимому, дефектами в сис-
теме окислительного фосфорилирования (Frank et al., 1978).
Злокачественная гипертермия, в свою очередь, часто соче-
тается с доминантно наследуемой миопатией, отличной от
болезни центрального стержня.
Семейный анализ сцепления заболевания с различны-
ми ДНК-маркерами позволил отнести мутантный ген CCD
(central core disease) в область 19q12-q13.2 (Haan etal., 1990;
Kaush et al., 1991). Было высказано предположение, что бо-
лезнь центрального стержня обусловлена мутациями в гене
рецептора-1 рианодина (RYR1 — ryanodine receptor 1), рас-
положенном в той же области генома (Mulley et al., 1993).
Это предположение было подтверждено при обнаружении
ряда мутаций в гене RYR1 у пациентов с болезнью централь-
ного стержня (Zhang et al., 1993; Quane et al., 1993). Таким
образом была доказана идентичность генов CCD и RYR1.
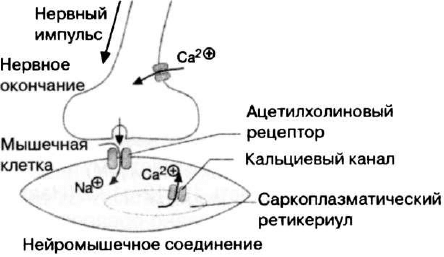
Рецептор рианодина — кальций-высвобождающий ка-
нал саркоплазматического ретикулума скелетных мышц (рис.
16). Рецептор рианодина необходим для развития и созрева-
ния мышц, а также его функции связаны с сокращением-рас-
слаблением миофибрилл. Трансгенные мыши с направленно
разрушенным геном Ryr1 погибают в перинатальном перио-
де и имеют значительные аномалии в развитии скелетных
мышц (Takeshima et al., 1994). ДНК-маркеры RYRI-гена ко-
сегерегируют с некоторыми формами злокачественной ги-
пертермии. Оказалось, что около 50% случаев злокачествен-
ной гипертермии являются аллельными вариантами болезни
центрального стержня и обусловлены различными миссенс-
мутациями в гене RYR1. Причем одна из этих мутаций —
R163C — встречалась у различных неродственных пациен-
131
тов, как с гипертермией, так и с болезнью центрального стерж-
ня, что указывает на возможность участия иных генетичес-
ких или средовых факторов в формировании подобной кли-
нической гетерогенности (Quane et al., 1993).
Рис. 16. Схема работы рецептора рианоцина
Некоторые пациенты с наследственной гипертрофиче-
ской кардиомиопатией, обусловленной мутациями в гене тя-
желой цепи р-миозина (MYH7), имеют выраженную мышеч-
ную слабость и гистологические аномалии, сходные с теми,
которые наблюдаются при болезни центрального стержня. У
одного из таких пациентов найдена миссенс-мутация — L908V
в гене MYH7, что доказывает возможность участия данного
гена в контроле некоторых форм болезни центрального стерж-
ня (Fananapazir et al., 1993).
д) Миотубулярная миопатия (MIM: 310400)
Данная форма врожденной миопатии отличается осо-
быми образованиями, выявляемыми гистологически и гис-
тохимически. Мышечные волокна в своей центральной час-
ти изменены, отличаются повышенной ферментативной ак-
тивностью и похожи на эмбриональные мышечные клетки. В
этом же отделе мышечного волокна обнаруживаются скоп-
ления мышечных ядер. Мышечные волокна уменьшены в раз-
132
мерах и отличаются повышенной активностью окислитель-
ных ферментов. Клинические проявления отмечаются с пер-
вых месяцев жизни в виде диффузной мышечной гипотонии,
атрофии и генерализованной слабости мышц. Глубокие ре-
флексы медленно снижаются. Течение, как правило, медленно
прогрессирующее, не исключена и стабилизация процесса.
Все это сближает клинические данные с таковыми при дру-
гих врожденных миопатиях. Однако следует отметить, что
заболевание имеет некоторые отличительные черты, способ-
ствующие клинической диагностике. К ним можно отнести
поражение глазодвигательных мышц, приводящее к косогла-
зию, и двусторонний, различной выраженности птоз. Миоту-
булярная миопатия — генетически гетерогенная группа кли-
нически сходных заболеваний.
В разных семьях прослеживается различный характер
наследования: аутосомно-доминантный, аутосомно-рецессив-
ный и Х-сцепленный рецессивный. В последнем случае за-
болевание характеризуется ранним началом и более тяже-
лым течением. При рождении больного ребенка наблюдает-
ся снижение или отсутствие спонтанных движений и наруше-
ние дыхания вследствие задержки или остановки нормаль-
ного развития мышечных волокон. Болезнь проявляется уже
в пренатальном периоде и часто сопровождается многово-
дием и слабым шевелением плода. У облигатных гетерозигот
часто наблюдаются выкидыши и мертворождения. большин-
ство пациентов погибают от дыхательной недостаточности в
возрасте 4—5 месяцев. Некоторые мальчики доживают до
взрослого периода за счет лучшей респирации после рожде-
ния. Причины подобной клинической гетерогенности неизве-
стны.
Описана косегрегация сцепленной с полом миотубуляр-
ной миопатии с маркерами области Xq28, в том числе с ге-
ном фактора VIII свертывания крови — F8C (Thomas et al.,
1987; 1990). Ген МТМ1 (myotubular myopathy 1) локализован
проксимальнее F8C (Lehesjoki etal., 1990), ближайшими флан-
кирующими микросателлитными маркерами являются
133
DXS304 и DXS305 (Dhal et al., 1994). Маркеры DXS455 и
DXS1684 наиболее информативны для диагностики аллель-
ного состояния гена МТМ1. Открытию гена сцепленной с по-
лом миотубулярной миопатии способствовало описание па-
циентов, несущих микроделеции в области локализации этих
маркеров (Dhal et al., 1995). Так, обнаружение делеции у де-
вочки с относительно благоприятным течением миотубуляр-
ной миопатии позволило ограничить вероятный район лока-
лизации гена МТМ1 600 тысячами пар оснований. Затем при
идентификации точек разрыва перекрывающихся микроде-
леции у двух мальчиков, характеризующихся миотубулярной
миопатией и аномалиями полового развития, этот район в
проксимальной области Xq28 дистальнее маркера DXS304
был сужен до 430 кб (Ни et al., 1996). Полученные результаты
ПОЗВОЛИЛИ использовать стратегию позиционного клонирова-
ния для изоляции гена МТМ1. Фрагменты ДНК из области
перекрывания двух таких микроделеции были клонированы
в дрожжевых и космидных векторах (Laporte et al., 1996). Для
нахождения транскрибируемых последовательностей в кло-
нированных ДНК использовали три различных подхода: (1)
отбор из тканеспецифических библиотек генов кДНКовых
последовательностей, гибридизующихся с геномными марке-
рами, сцепленными с геном МТМ1, (2) улавливание экзонов
и (3) частичное секвенирование клонированных геномных
последовательностей с последующим компьютерным поиском
кодирующих областей. В результате было отобрано пять эк-
зонов и шесть кДНК-клонов, содержащих частично перекры-
вающиеся фрагменты ДНК. Кроме того, три района секвени-
рованных космидных последовательностей соответствовали
кодирующим областям ДНК. При сопоставлении мест лока-
лизации этих элементов был идентифицирован неизвестный
ранее ген — CG1, занимающий протяженную центральную
часть области перекрывания двух микроделеции. Этот ген
состоит из 7 экзонов и преимущественно экспрессируется в
скелетных мышцах; размер мРНК составляет 4.6 кб. CG1
рассматривался как наиболее вероятный кандидат для МТМ1.
134
Однако предположение не нашло подтверждения, так как ни
у одного из 28 пациентов с Х-сцепленной миотубулярной
миопатией мутаций в кодирующей части гена CG1 обнару-
жено не было. Возможно, этот ген является кандидатным для
каких-то других наследственных заболеваний, обусловлен-
ных повреждением области Xq28, таких как синдром Вайс-
мана, Х-сцепленная умственная отсталость (MRX3) или ото-
палато-дигитальный синдром (OPD).
Второй идентифицированный ген — CG2, состоящий
из 15 экзонов, отсутствовал у одного из пациентов с мик-
роделециями и был частично делетирован у другого. До-
полнительный скрининг 12 тканеспецифических библиотек
и компьютерный анализ базы данных EST-последователь-
ностей позволили идентифицировать фрагмент кДНК раз-
мером в 3.4 кб с сигналом полиаденилирования и откры-
той рамкой считывания. Этот фрагмент кДНК способен
кодировать полипептидную цепь, состоящую из 621 ами-
нокислоты. Причем в клонах кДНК, изолированных соот-
ветственно из библиотеки печени и скелетных мышц, при-
сутствовали альтернативные сайты полиаденилирования.
Оказалось, что ген CG2 идентичен МТМ1, так как были
обнаружены различные мутантные аллели этого локуса у
пациентов с Х-сцепленной миотубулярной миопатией. На-
ряду с миссенс-мутациями описаны небольшие делеций и
инсерции, приводящие к сдвигу рамки считывания. Иссле-
дование 40% кодирующей области гена МТМ1 позволило
идентифицировать мутации у 12% пациентов.
Продукт гена CG2, получивший название миотубулярин,
имеет высокую гомологию по последовательности с одним
из белков Saccharamyces cerevisiae и Caenorhabditis elegans
(53% и 56% соответственно). Кроме того, анализ базы дан-
ных EST-последовательностей показал, что в геноме чело-
века существует по крайней мере восемь других не описан-
ных ранее генов, высокогомологичных МТМ1. Определена
хромосомная локализация и характер экспрессии большин-
ства из них (Laporte et al., 1997). Один из этих генов локали-
135
зован дистальнее МТМ1 в Xq28. Миотубулярин содержит рай-
он (РТР), сходный с активным центром тирозинфосфатаз. Эти
белки вовлечены в регуляцию путей сигнальной трансдукции,
участвующих в контроле процессов роста, пролиферации и
дифференцировки клеток. Оказалось, что миотубулярин спо-
собен гидролизовать синтетический аналог тирозинфосфата
в реакции in vitro, ингибируемой ортованадатом. По-видимо-
му, миотубулярин является членом нового семейства тиро-
зинфосфатаз (DSP), обладающих двойной специфичностью
как по отношению к тирозину, так и по отношению к серину.
Дефекты этого белка сопровождаются нарушениями нормаль-
ного процесса созревания мышц, что и приводит к развитию
миотубулярной миопатии. Другие гомологи миотубулярина не
имеют функционального РТР-активного сайта. МТМ1 принад-
лежит к семейству высококонсервативных генов, обнаружен-
ных не только в геноме млекопитающих, но и у различных
видов дрожжей, а также у дрозофилы (Laporte et al., 1997).
Сравнение этого ряда генов позволяет реконструировать
филогенетическое дерево и определять консервативные ос-
татки, наиболее существенные для выполнения общих функ-
ций соответствующих белков.
Прямое секвенирование 92% кодирующей части МТМ1
гена у мальчиков с миотубулярной миопатией, диагностиро-
ванной на основе гистологического анализа биоптатов мышц,
позволило идентифицировать 26 мутаций, 50% которых были
локализованы в экзонах 4 и 12 (de Gouyon et al., 1997). У
большинства пациентов обнаруживаются точковые мутации,
4 из которых нарушают процесс сплайсинга. Одна из мис-
сенс-мутаций затрагивает высококонсервативную аминокис-
лоту, присутствующую во всех гомологичных белках других
исследованных видов начиная с дрозофилы. У остальных па-
циентов найдены делеции размером от нескольких нукле-
отидов до 2—6 экзонов. Три мутации встретились более чем
у одного пациента. В параллельном исследовании было про-
ведено скринирование всей кодирующей области МТМ1 гена
методом SSCP-анализа у 85 неродственных пациентов
136
(Laporte et al., 1997). В 55 случаях удалось идентифициро-
вать мутантные аллели. Большие делеций обнаружены толь-
ко у трех пациентов. 5 точковых мутаций найдены у многих
пациентов и, по-видимому, они обьясняют 27% всех случаев
заболевания. Более половины мутаций нарушают фермен-
тативную активность миотубулярина, вследствие преждевре-
менной терминации трансляции либо в результате аминокис-
лотных замен в фосфатазном домене белка. Остальные мис-
сенс-мутации кластерированы в двух консервативных райо-
нах миотубулярина, что является указанием на присутствие
в этом белке других функциональных доменов, не связан-
ных непосредственно с реакцией фосфорилирования.
е) Миопатия Бетлема (MIM: 158810)
Генетически гетерогенная группа врожденных миопа-
тии с аутосомно-доминантным типом наследования. Заболе-
вание характеризуется врожденной мышечной гипотонией,
укладывающейся в картину синдрома «вялого ребенка». В
последующем наблюдается медленно прогрессирующая ат-
рофия мышц с возможным развитием кардиомиопатий и
множественных контрактур суставов (Bethlem, van Wijn-
gaarden, 1976; Arts et al., 1978). В биоптатах мышц наблюда-
ется атрофия волокон I типа.
При генетическом анализе датских семей с данной фор-
мой миопатии обнаружено сцепление локуса, ответственно-
го за развитие заболевания, с областью 21q22.3, в которой
расположен кластер генов, кодирующих а1 и ос2 цепи колла-
гена типа VI (Jobsis et al., 1995; Jobsis et al., 1996a). В боль-
шой франко-канадской семье, в которой одновременно на-
блюдали 19 пациентов, показано сцепление исследуемого
локуса с маркерами D2S336 и D2S395, локализованными в
17-сМ области 2q37, в которой расположен ген аЗ цепи кол-
лагена того же типа VI (Speer et al., 1996). В настоящее вре-
мя идентифицировано19 типов коллагенов, состоящих из
более чем 30 полипептидных цепей, кодируемых разными
генами. Генетические дефекты некоторых типов коллагенов
137
связаны с рядом моногенных заболеваний, таких как несо-
вершенный остеогенез, синдром Элерса-Данлоса (тип IV и
VII), синдром Альпорта, некоторые формы хондродисплазий.
При этих заболеваниях нарушаются процессы посттрансля-
ционного созревания либо протеолитического расщепления
коллагеновых цепей, что приводит к неправильному спари-
ванию мономеров либо к образованию мультимерных ансам-
блей большего размера.
Зрелые молекулы коллагеновых белков состоят из трех
равномерно скрученных полипептидных а-цепей, образую-
щих структуру трехгранного стержня. Разные типы коллаге-
нов могут быть образованы либо тремя одинаковыми а-це-
пями, либо двумя или тремя различными полипептидами в
соотношении 2:1 или 1:1:1 соответственно. Любая а-цепь со-
держит коллагеновый домен, на всем протяжении которого
за исключением короткого С-терминального участка каждая
третья аминокислота является глицином (Gly). Таким обра-
зом, молекулярная формула коллагенового домена может
быть записана как (Gly-X-Y)n, где X и Y — аминокислоты не-
Gly типа. Различные коллагеновые а-цепи различаются по
количеству (Gly-X-Y) мотивов в коллагеновом домене и по
конкретному содержанию аминокислот в X и Y положениях.
Некоторые коллагены содержат также неколлагеновые доме-
ны. Присутствие глицина, самой небольшой из аминокислот,
в каждом третьем положении коллагеновых полипептидных
цепей существенно для их правильного скручивания в трой-
ную спираль, так как глицин при этом занимает ограничен-
ное пространство в центре триплекса. Поэтому любые мута-
ции, приводящие к замене глицина на другую аминокислоту,
будут сопровождаться локальным нарушением структуры
тройной спирали. Все коллагеновые молекулы способны об-
разовывать супрамолекулярные агрегаты, частично стаби-
лизированные за счет взаимодейсвий между триплексными
доменами. Коллагены типов l,ll, III, V, VI и XI формируют фи-
бриллыдо есть являются фибриллярными коллагенами. Кол-
лагены типа IV агрегируются, образуя слои базальных мемб-
138
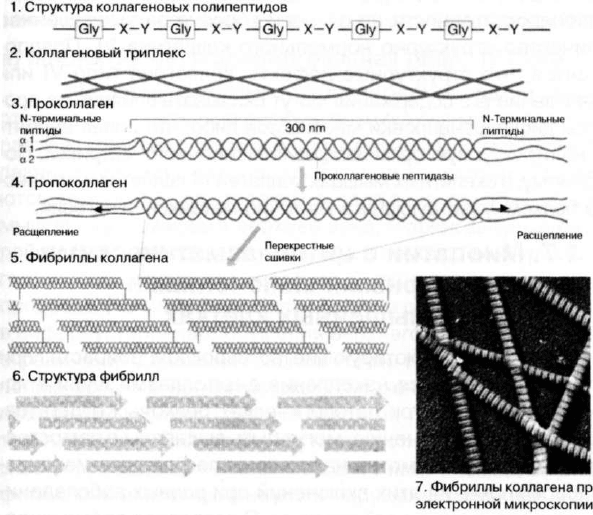
ран, коллагены VIII формируют десцеметовую мембрану и
коллагены типа VII участвуют в образовании латерально аг-
регированных антипараллельных димеров, являющихся глав-
ным компонентом опорных фибрилл. На рис. 17 изображена
структура коллагена. Предполагается, что среди большого се-
мейства коллагенов широко экспрессирующийся микрофиб-
риллярный коллаген VI типа выполняет роль моста между
клетками и внеклеточным матриксом. Этот тип коллагена при-
сутствует не только в эндомизии и перимизии скелетных
мышц, но и во многих других тканях.
Рис. 17. Структура коллагена
При секвенировании амплифицированных фрагментов
кодирующих областей генов COL6A1 (collagen 6 alpha 1) и
COL6A2, полученных от пациентов с миопатией Бетлема,
139
ассоциированной с областью 21q22.3, в трех семьях были
обнаружены две гетерозиготные миссенс-мутации. Одна со-
ответственно в гене COL6A1, а другая однотипная мутация в
двух семьях — в гене COL6A2 (Jobsis et al., 1996b). Обе му-
тации разрушали Gly-X-Y мотив в стержневом домене колла-
гена путем замещения глицина либо на валин, либо на се-
рии. Гетерозиготное состояние по делеции одного нуклеоти-
да в гене COL6A1 также приводит к развитию миопатии Бет-
лема (Lamandiet al., 1998). Эта мутация сопровождается преж-
девременной терминацией трансляции, при этом мутантная
мРНК оказывается нестабильной и почти полностью отсутст-
вует в фибробластах и в скелетных мышцах больных. При
гаплонедостаточности по а1-цепи образуется уменьшенное
количество структурно нормального коллагена VI. Предпо-
лагается, что структурные дефекты коллагена типа VI или
уменьшение его содержания могут оказывать влияние на про-
цессы дифференцировки миобластов либо, что более вероят-
но, на структурную организацию внеклеточного матрикса. По-
видимому, в скелетных мышцах коллаген VI является критичес-
ким белком, участвующим в клеточно-матриксной адгезии.
1.7. Миопатии с цитоплазматическими
и ядерными включениями
в мышечных клетках
Миопатии, дебютирующие во взрослом возрасте, при
которых наблюдается накопление в цитоплазме и/или в яд-
рах мышечных клеток патологических белковых агрегатов,
также, по нашему мнению, могут быть выделены в самостоя-
тельную группу, несмотря на то что молекулярные механиз-
мы формирования этих включений при разных заболевани-
ях могут значительно различаться. В последнее время широ-
ко обсуждается роль подобных образований в развитии ней-
родегенеративных процессов при таких разных заболевани-
ях, как болезнь Альцгеймера, некоторые семейные формы
болезни Паркинсона и бокового амиотрофического склеро-
140