Фёршт Э. Структура и механизм действия ферментов
Подождите немного. Документ загружается.


СТРУКТУРА
И МЕХАНИЗМ
ДЕЙСТВИЯ
ОТДЕЛЬНЫХ
ФЕРМЕНТОВ
351
первой стадией которого является связывание кофермента, а
лимитирующей стадией — диссоциация комплекса фермент —
NADH
[10, 16, 17]. И теория переходного состояния, и данные
стационарной
кинетики показывают, что образующийся комп-
лекс фермент—NAD+ изомеризуется [17, 18]. Что касается
обратной реакции, то при восстановлении ароматических альде-
гидов лимитирующей стадией является стадия диссоциации
комплекса
между
ферментом и спиртом [17, 19],
тогда
как вос-
становление ацетальдегида лимитируется химической стадией
(перенос
гидрид-иона).
Комплексы
между
ферментом и продуктом для алкогольде-
гидрогеназы дрожжей диссоциируют довольно быстро, так что
лимитирующими являются стадии химического превращения
[20].
Это позволяет измерить кинетические изотопные эффекты
для указанных стадий с помощью стационарной кинетики.
Установлено, что окисление дейтерированных спиртов RCD
2
OH
и
восстановление бензальдегидов дейтерированным NADH (т. е.
NADD)
протекают значительно медленнее соответствующих ре-
акций
с участием немеченых соединений
(&H/&D
= 3—5) [14,
20].
Это показывает, что перенос гидрид-иона (или дейтерид-
иона)
осуществляется в
ходе
лимитирующей стадии реакции.
Измерение
константы скорости переноса гидрид-иона в
случае
алкогольдегидрогеназы печени лошади с помощью предстацио-
нарной
кинетики выявило наличие аналогичных изотопных эф-
фектов
[21, 22].
Основной
момент, остающийся неясным для рассматривае-
мого механизма, касается координационного числа иона
Zn
2+
и
состояния
ионизации его лигандов. Соответствующие данные
были получены при исследовании зависимости реакционной спо-
собности фермента от его структуры и рН-зависимости скорости
реакции.
Установлено, что k
ca
t для реакции окисления спиртов
алкогольдегидрогеназой дрожжей и
fe
ca
t
для реакции восста-
новления
бензальдегидов алкогольдегидрогеназой печени слабо
зависят от способности реагирующих атомов принимать или
отдавать электроны [14, 15, 20, 23, 24]. Возможно, это обуслов-
лено тем, что перенос гидрид-иона осуществляется по механизму
общего кислотно-основного катализа (т. е. образующийся при
переносе гидрид-иона заряд нейтрализуется синхронным пере-
носом
протона от кислородного атома субстрата или на этот
атом).
Однако никакой боковой цепи, которая находилась бы
достаточно близко к
субстрату,
чтобы выполнять каталитиче-
скую функцию, не обнаружено. Было высказано предположение,
что карбонильная группа субстрата присоединяется не к самому
иону цинка, а к связанной с ним молекуле воды [25]. Хотя это
согласуется с предположением о наличии общего кислотно-основ-
ного катализа, в котором связанная с цинком вода играет роль

352
ГЛАВА
12
общей кислоты, а связанный с цинком гидроксил-ион — общего
основания,
геометрия указанного комплекса, по-видимому, про-
тиворечит данным рентгеноструктурного анализа [26]. Согласно
другой
гипотезе, связанная с цинком молекула воды выполняет
роль катализатора, а
субстрат
присоединяется координационной
связью непосредственно к атому цинка. При этом предполагает-
ся,
что связывание субстрата с цинком не приводит к вытесне-
нию
молекулы воды, а увеличивает координационное число цин-
ка
до пяти [уравнение
(12.4)]
[15].
+
/->А NAD-
NAD
+
/H
\
R
Для выяснения состояния ионизации каталитической группы
была исследована рН-зависимость скорости ферментативной ре-
акции.
При окислении n-метилбензилового спирта, катализируе-
мом алькогольдегидрогеназой дрожжей, k
c
m возрастает при
увеличении рН и определяется состоянием ионизации осно'вной
формы
группы с р/Са = 8,25,
тогда
как при восстановлении
ацетальдегида параметр
kcat/Км.
возрастает при уменьшении
рН
и определяется состоянием ионизации кислой формы группы
с р/(
а
=8,25 [27]. Это не противоречит механизму (12.4), по-
скольку связанные с цинком молекулы воды в модельных со-
единениях ионизируются именно в этой области рН [28]. Значи-
тельно
труднее
интерпретировать рН-зависимость k
ca
t для ре-
акций,
катализируемых нативной алкогольдегидрогеназой из
печени
лошади. Скорость восстановления альдегидов мало из-
меняется
в диапазоне рН от 6 до 10 [15], что можно рассматри-
вать как довод в пользу выполнения механизма
(12.3)
[29].
Если
имеет место механизм (12.4), то р/С
а
связанной с цинком
молекулы воды в тройном комплексе с NADH и альдегидом
должен быть относительно высоким, а рК
а
связанного с цинком
гидроксильного иона в тройном комплексе с NAD+ и альдеги-
дом— относительно низким [15]. Аналогичным образом, если
справедлив механизм (12.3), то связанный с цинком алкоголят-
ион
должен иметь необычно низкое значение р/(
а
.
Вопрос о том, обладают ли реакционной способностью все
активные центры молекулы алкогольдегидрогеназы или полови-
на
их, остается открытым. Хотя этот фермент связывает два
моля
NAD+ с одинаковым сродством, считается, что в катализе
участвует
только один активный центр.

СТРУКТУРА
И МЕХАНИЗМ ДЕЙСТВИЯ ОТДЕЛЬНЫХ ФЕРМЕНТОВ 353
2. L-лактатдегидрогеназа [4, 6, 32]
L-лактатдегидрогеназа катализирует обратимое окисление
L-лактата до пирувата с использованием в качестве кофермента
NAD+:
СНз СНз
-OH +
NAD
+
=f=fc
C=O +
NADH
+ H
+
(12.5)
I I
COJ СОГ
H—С—С
•>2
Этот фермент, выделенный из многих источников, представ-
ляет собой тетрамер с мол. весом 140 000. Он может существо*
вать в
двух
формах: Н
4
, преобладающей в сердечной мышце, и
М
4
, преобладающей в скелетной мышце [33, 34]. Эти формы
называются
изоферментами
— разными молекулярными форма-
ми
одного и того же фермента. Аминокислотный состав форм
М.4 и Н
4
существенно различается; кинетические свойства изо-
ферментов также неодинаковы. Несмотря на это, свойства цент-
ров ассоциации субъединиц весьма близки, поскольку вероят-
ность образования таких гибридных форм, как М
3
Н, М
2
Нг и МН
3
,
определяется чисто статистическими факторами [35]. В
ходе
каталитической реакции субъединицы
никак
не взаимодейству-
ют
между
собой, так что кинетические свойства, например,
формы
Н
3
М и смеси форм Н
4
и М
4
в соотношении 3 : 1 идентичны.
Кристаллическая
структура
апофермента, выделенного из аку-
лы,
была установлена с разрешением 2,0 А, а его комплекса с
аддуктом NAD—пируват — с разрешением 2,8 А [36]. Молеку-
ла фермента симметрична, а субъединицы структурно эквива-
лентны.
а.
Структура
фермент-субстратного
комплекса
Структура тройных комплексов была определена из кри-
сталлографических исследований [37] связывания
аддукта
NAD—пируват, который можно рассматривать как аналог свя-
занных ковалентной связью пирувата и NADH [38]:
со;
354
ГЛАВА
12
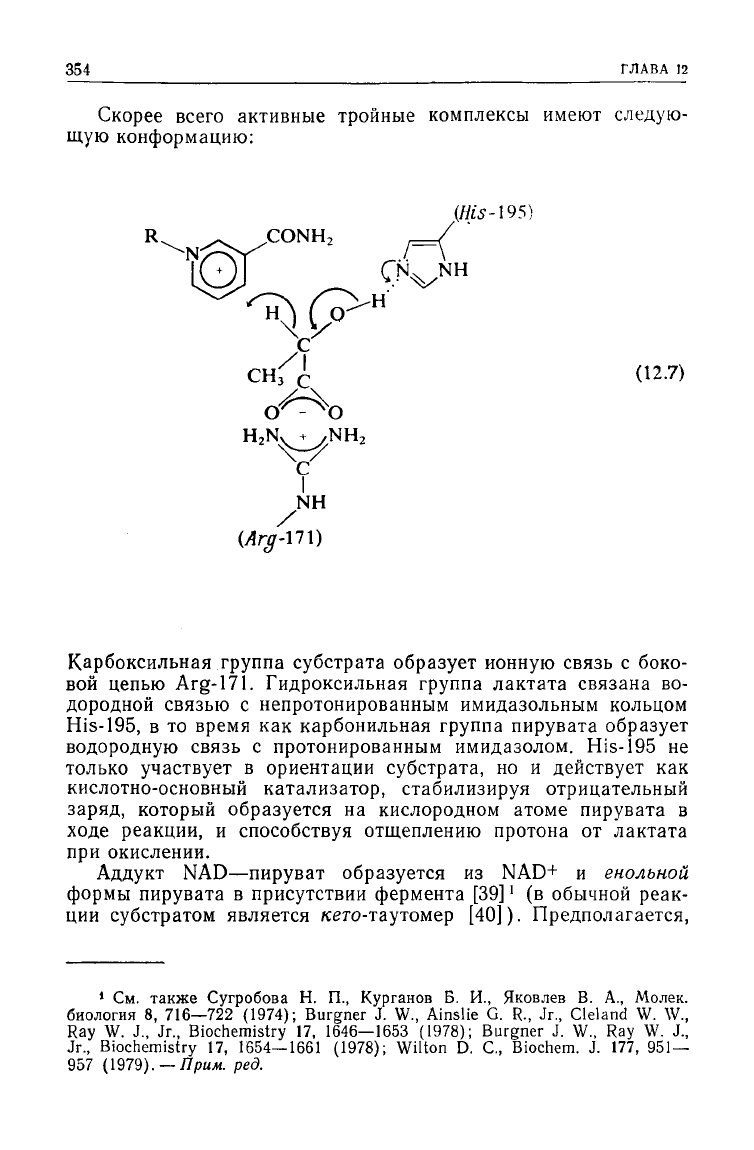
Скорее всего активные тройные комплексы имеют следую-
щую конформацию:
(12.7)
Карбоксильная
группа субстрата образует ионную связь с боко-
вой
цепью
Arg-171.
Гидроксильная группа лактата связана во-
дородной связью с непротонированным имидазольным кольцом
His-195, в то время как карбонильная группа пирувата образует
водородную связь с протонированным имидазолом. His-195 не
только
участвует
в ориентации субстрата, но и действует как
кислотно-основный
катализатор, стабилизируя отрицательный
заряд, который образуется на кислородном атоме пирувата в
ходе
реакции, и способствуя отщеплению протона от лактата
при
окислении.
Аддукт
NAD—пируват образуется из NAD+ и
енольной
формы
пирувата в присутствии фермента [39]' (в обычной реак-
ции
субстратом является /сего-таутомер [40]). Предполагается,
1
См. также Сугробова Н. П., Курганов Б. И., Яковлев В. А., Молек.
биология
8,
716—722
(1974); Burgner J. W.,
Ainslie
G. R., Jr., Cleland W. W.,
Ray W. J., Jr., Biochemistry 17,
1646—1653
(1978); Burgner J. W., Ray W. J,
Jr., Biochemistry 17,
1654—1661
(1978); Wilton D. C, Biochem. J. 177, 951—-
957 (1979). —
Прим.
ред.
СТРУКТУРА
И
МЕХАНИЗМ
ДЕЙСТВИЯ
ОТДЕЛЬНЫХ
ФЕРМЕНТОВ
355
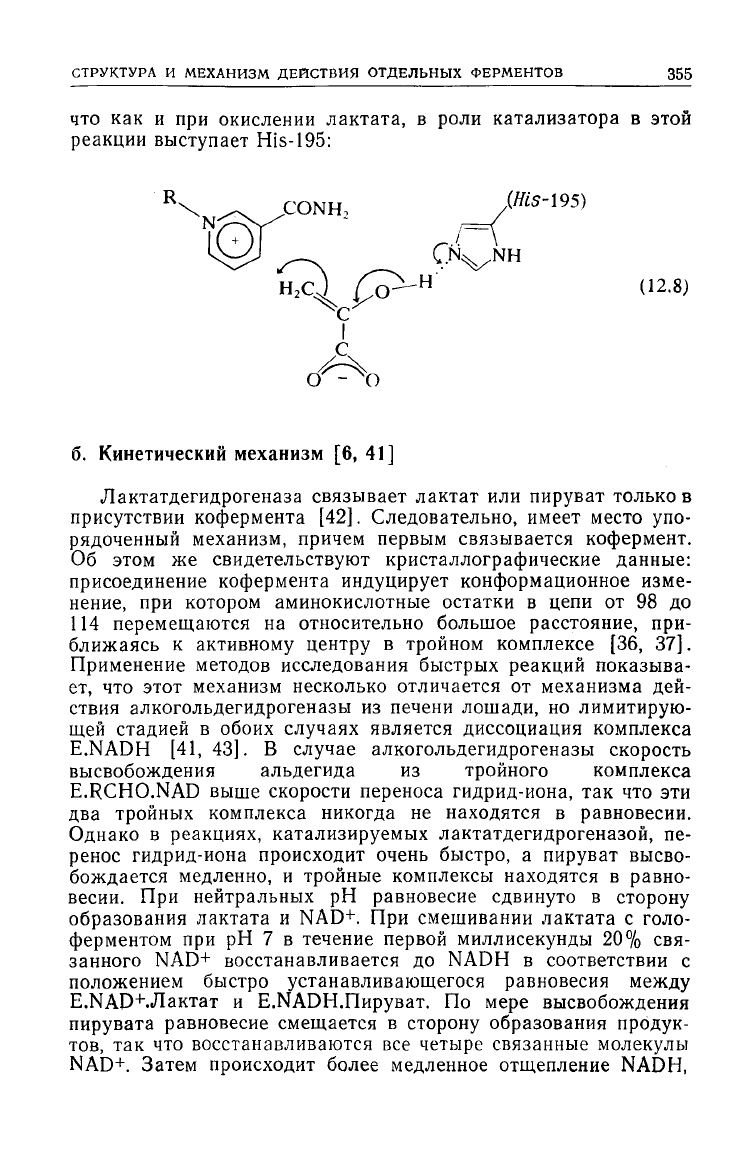
что как и при окислении лактата, в роли катализатора в этой
реакции
выступает His-195:
(HiS-195)
(12.8)
б. Кинетический механизм [6, 41]
Лактатдегидрогеназа связывает лактат или пируват только в
присутствии кофермента [42]. Следовательно, имеет место упо-
рядоченный
механизм, причем первым связывается кофермент.
Об этом же
свидетельствуют
кристаллографические данные:
присоединение
кофермента индуцирует конформационное изме-
нение,
при котором аминокислотные остатки в цепи от 98 до
114 перемещаются на относительно большое расстояние, при-
ближаясь к активному центру в тройном комплексе [36, 37].
Применение
методов исследования быстрых реакций показыва-
ет, что этот механизм несколько отличается от механизма дей-
ствия алкогольдегидрогеназы из печени лошади, но лимитирую-
щей
стадией в обоих
случаях
является диссоциация комплекса
E.NADH
[41, 43]. В
случае
алкогольдегидрогеназы скорость
высвобождения альдегида из тройного комплекса
E.RCHO.NAD
выше скорости переноса гидрид-иона, так что эти
два тройных комплекса никогда не находятся в равновесии.
Однако в реакциях, катализируемых лактатдегидрогеназой, пе-
ренос гидрид-иона происходит очень быстро, а пируват высво-
бождается медленно, и тройные комплексы находятся в равно-
весии.
При нейтральных рН равновесие сдвинуто в сторону
образования
лактата и
NAD+.
При смешивании лактата с голо-
ферментом при рН 7 в течение первой миллисекунды 20% свя-
занного
NAD+ восстанавливается до NADH в соответствии с
положением быстро устанавливающегося равновесия
между
E.NAD+.Лактат
и E.NADH.Пируват. По мере высвобождения
пирувата равновесие смещается в сторону образования продук-
тов, так что восстанавливаются все четыре связанные молекулы
NAD+.
Затем происходит более медленное отщепление NADH,

366
ГЛАВА
12
являющееся лимитирующей стадией
в
стационарном состоянии:
Быстро
Медленно
E.NAD+.Лактат
t
-» E.NADH.Пируват ->- >
Пируват
Самая
медленная стадия
-^> E.NADH
^ * Е.
(12.9)
NADH
Такое поведение можно
по
ошибке объяснить
тем, что
реакци*
онной
способностью обладает половина активных центров
в мо-
лекуле, однако
все
четыре центра, по-видимому, являются неза-
висимыми
[6, 41, 43].
Связывание коферментов также происхо*
дит независимо
с
каждым центром.
Играющий
большую роль
в
катализе остаток
His-195
обла-
дает
необычной реакционной способностью
в
отношении диэтил-
пирокарбоната.
Это
дает
возможность определить
р/С
а
(=6,7)
как
апо-, так и
голофермента непосредственно
из
рН-зависимо-
сти скорости модификации
[44].
Имеются данные
о том, что
лактат предпочтительно связывается
с
голоферментом, несущим
неионизированный
гистидин,
тогда
как
пируват
— с
комплексом
E.NADH,
гистидин которого протонирован.
3. Малатдегидрогеназа
[7]
Малатдегидрогеназа катализирует обратимое окисление
ма-
лата
до
оксалоацетата
с
использованием
в
качестве кофермента
NAD+:
СО
2
"
COJ
Н—С—OH
+
NAD
+
ч=±
C=O
+
NADH
+
H*
(12.10)
I
I
I
I
CH
2
COJ
СН
2
СО
2
"
Кристаллическая
структура
растворимого (цитоплазматическо-
го) фермента установлена
с
разрешением
2,5 А [45].
Карта
электронной
плотности
еще не
соотнесена
с
аминокислотной
последовательностью,
но, как
указывалось
в гл. 1,
укладка
по-
липептидной
цепи фермента
в
принципе аналогична укладке
цепи
лактатдегидрогеназы, хотя малатдегидрогеназа является
всего лишь димером
с мол.
весом
70 000.
Вероятно, механизм
действия обоих ферментов одинаков, поскольку малатдегидро-
геназа также содержит важный
для
катализа остаток гистиди-
на,
который может модифицироваться диэтилпирокарбонатом
[46].
Два
моля NADH
(или
NAD+)
связываются
с
одинаковым
сродством
[47].
Кажущаяся отрицательная кооперативность
при
связывании
кофермента, обнаруженная
для
одного
из
препара-
тов фермента, может быть обусловлена
тем, что на
самом
деле
в
препарате присутствовали
две
формы
[48, 49].

СТРУКТУРА
И
МЕХАНИЗМ
ДЕЙСТВИЯ
ОТДЕЛЬНЫХ
ФЕРМЕНТОВ 357
4.
Глицеральдегид-3-фосфат—дегидрогеназа
[50]
Глицеральдегид-3-фосфат—дегидрогеназа, тетрамерный фер-
мент с мол. весом 150 000, состоит из четырех идентичных цепей;
он
катализирует обратимое окислительное фосфорилирование
глицеральдегид-3-фосфата до 1,3-дифосфоглицерата с использо*
ванием
в качестве кофермента
NAD+:
с
с
Н—С—ОН
+
NAD
+
+
НРОГ
^=fc
Н—С—OH
+
NADH
+ H* (12.11)
CHjOPOf CHjOPOf
Эта реакция протекает через несколько стадий. Адекватность
механизма окислительного фосфорилирования [реакции
(12.12)
—(12.16)], предложенного в 1953 г. [51], подтвержда-
ется всесторонними исследованиями с привлечением данных
предстационарной [52] и стационарной [53] кинетики;
ОН
NAD
+
.E—
SH
+ RCHO
*=*=
NAD
+
.E—S—C—R
(12.12)
NAD
+
.E—S—C—R
*=±
NADH.E—S—С
+ Н
+
(12.13)
N
NADH.E—
S—C"^
=<=-
v
-
E—
S—o/ +
NADH
(12 14)
4
R
4
R
E—
S—QT
+NAD
+
^=fc
NAD
+
.E—S—C*^
(12.15)
4
\
3
/ОРОГ
NAD+.E—
S—С
+НРОГ
ч=±
NAD
+
.E—SH
+ RC (12.16)
В ферменте имеется реакционноспособныи остаток цистеина,
который легко ацилируется ацилфосфатами с образованием тио-
эфира
[обратное направление реакции
(12.16)]
[54]. Первая ста-
дия
в приведенной последовательности реакций состоит в обра-
зовании
полутиоацеталя
между
цистеином и субстратом. На
этой
стадии происходит превращение карбонильной группы,
трудно поддающейся прямому окислению, в спиртовую группу,
легко дегидрирующуюся обычным путем [реакция (12.13)].

358
ГЛАВА
12
Образовавшийся в
ходе
реакции
(12.13)
тиоэфир взаимодей-
ствует
с ортофосфатом, давая ацилфосфат (12.16). Однако
перенос ацильной группы происходит очень медленно до тех пор,
пока
NAD+ не присоединится к ферменту [55, 56]. Поэтому
обязательным этапом в последовательности приведенных выше
реакций
является замещение NADH на NAD+ в
ходе
реакций
(12.14)
и (12.15). Интересно, что диссоциация комплекса ацил-
фермента с NADH
(12.14)
при насыщающих концентрациях
реагента и высоком рН является лимитирующей стадией данной
последовательности реакций [55]. Следствием замещения
NADH
на NAD+ до высвобождения ацилфосфата является то,
что свободный апофермент не принимает участие в реакции.
Кроме
того, поскольку ацилирование фермента дифосфатом ак-
тивируется
NAD+,
голофермент инициирует восстановительное
дефосфорилирование 1,3-дифосфоглицерата.
Комплексы
Михаэлиса голофермента с альдегидом или ди-
фосфоглицератом и ацилфермента с ортофосфатом в эту
схему
не
включены, поскольку их константы диссоциации слишком
высоки,
чтобы могло происходить накопление комплексов.
а.
Структура
фермент-субстратных
комплексов
Кристаллическая
структура
голоферментов, выделенных из
омара [57] и
Bacillus
sterothermophilus
[58], была установлена
с разрешением 2,9 и 2,7 А соответственно, а
структура
голофер-
мента человека [59] и бактериального апофермента [58] — с
низким
разрешением. Структура фермент-субстратных комплек-
сов определена с помощью построения моделей для фермента,
выделенного из омара, и бактериального фермента [57, 58].
Далее мы
всюду
будем
рассматривать данные, касающиеся
только бактериального фермента.
В активном центре бактериального фермента, полученного в
кристаллической форме из раствора сульфата аммония, выявле-
ны
два центра связывания сульфат-ионов (рис. 12.1) [60]. Ра-
зумную с точки зрения химии и стереохимии
схему
реакции
можно построить, предположив, что один из этих
двух
центров
представляет собой центр связывания фосфатного остатка суб-
страта, а
другой
— центр связывания фосфатного остатка, при-
нимающего участие в реакции деацилирования (12.16). Когда
3-фосфат образует водородные связи с гидроксильной группой
Thr-179, положительно заряженной боковой цепью
Arg-231
и
2'-гидроксильной группой рибозного кольца, присоединенного к
никотинамиду
NAD+,
альдегидная группа субстрата может об-
разовывать с остатком
Cys-149
полутиоацеталь (рис.
12.1).
За-
тем гидроксильная группа субстрата в положении С-2 может
образовать водородную связь с Ser-148, а гидроксильная группа
СТРУКТУРА
И
МЕХАНИЗМ
ДЕЙСТВИЯ
ОТДЕЛЬНЫХ
ФЕРМЕНТОВ
359
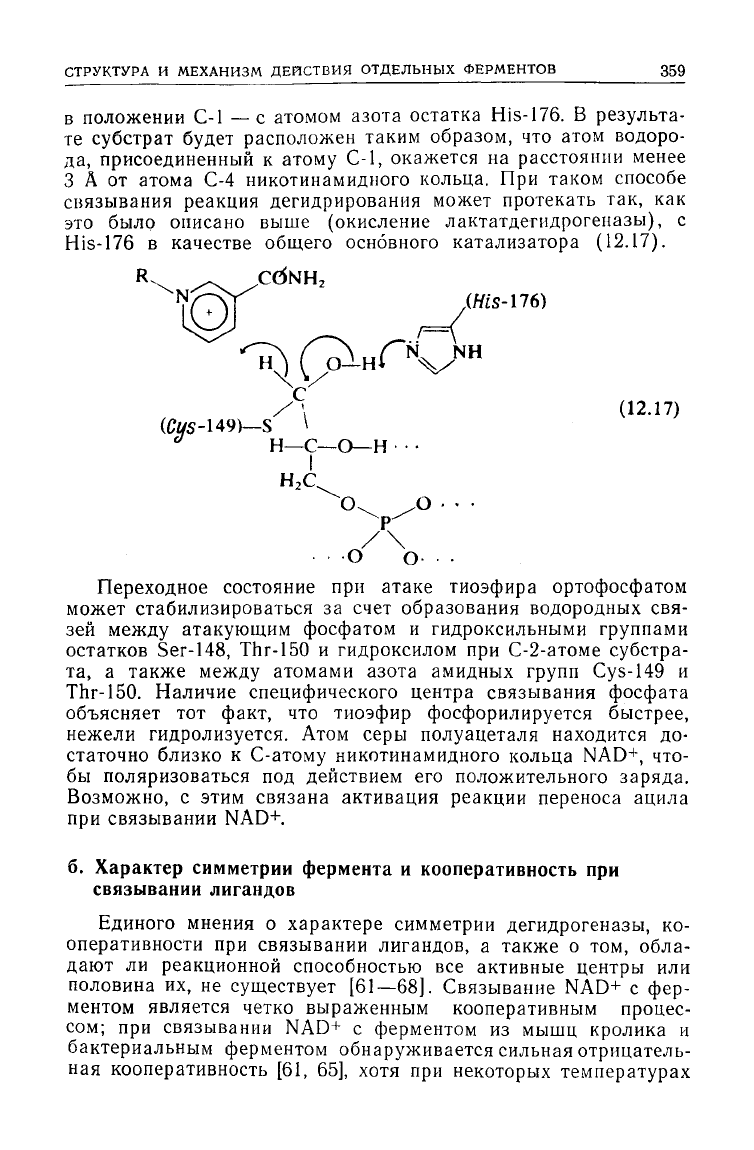
в
положении С-1 —с атомом азота остатка His-176. В результа-
те субстрат
будет
расположен таким образом, что атом водоро-
да, присоединенный к атому С-1, окажется на расстоянии менее
3 А от атома С-4 никотинамидного кольца. При таком способе
связывания
реакция дегидрирования может протекать так, как
это
было описано выше (окисление лактатдегидрогеназы), с
His-176 в качестве общего основного катализатора (12.17).
AHis-nS)
/\
(12.17)
(ClfS-149)—S
\
d
Н—С—О—Н
Н
2
С
о
о •
Переходное состояние при атаке тиоэфира ортофосфатом
может стабилизироваться за счет образования водородных свя-
зей
между атакующим фосфатом и гидроксильными группами
остатков Ser-148, Thr-150 и гидроксилом при С-2-атоме субстра-
та, а также между атомами азота амидных групп
Cys-149
и
Thr-150. Наличие специфического центра связывания фосфата
объясняет тот факт, что тиозфир фосфорилируется быстрее,
нежели гидролизуется.
Атом
серы полуацеталя находится до-
статочно близко к С-атому никотинамидного кольца
NAD+,
что-
бы поляризоваться под действием его положительного заряда.
Возможно, с этим связана активация реакции переноса ацила
при
связывании
NAD+.
б.
Характер
симметрии
фермента
и
кооперативность
при
связывании
лигандов
Единого мнения о характере симметрии дегидрогеназы, ко-
оперативное™ при связывании лигандов, а также о том, обла-
дают ли реакционной способностью все активные центры или
половина
их, не существует [61—68]. Связывание NAD+ с фер-
ментом является четко выраженным кооперативным процес-
сом;
при связывании NAD+ с ферментом из мышц кролика и
бактериальным ферментом обнаруживается сильная отрицатель-
ная
кооперативность [61, 65], хотя при некоторых температурах

360
ГЛАВА
12
присоединение
NAD+ к дегидрогеиазе дрожжей характеризует-
ся
положительной кооперативностью [69]. С
другой
стороны,
присоединение
глицеральдегид-3-фосфата ко всем четырем
субъединицам происходит независимо [68]. Наличие реакцион-
ной
способности у половины активных центров установлено
только в
случае
искусственных субстратов; например,
1,3-ди-
фосфоглицерат ацилирует все четыре реакционноспособных
остатка цистеина с одной и той же константой скорости [55, 63,
65, 68]. На основании данных ряда кинетических работ и ре-
зультатов исследования связывания было высказано предполо-
жение,
что эта дегидрогеназа
существует
в виде «димера диме-
ров», состоящего из
двух
пар структурно различающихся
субъединиц [67, 70]. Анализ карты электронной плотности кри-
сталлического голофермента из омара, полученной при высоком
разрешении,
подтверждает эту точку зрения [57], однако более
поздние исследования бактериального фермента четко показы-
вают, что все четыре субъединицы структурно идентичны и дан-
ный
фермент имеет симметрию 222 [58]. Бактериальный апо-
фермент обладает такой же симметрией, хотя исследования
проводились только при низком разрешении [58]. Сравнитель-
ный
анализ свойств бактериальных апо- и голоферментов пока-
зывает, что связывание NAD+ вызывает существенное смещение
домена, на котором связывается кофермент, и это приводит к
уменьшению объема молекулы. Структурные предпосылки отри-
цательной кооперативности пока не выявлены.
5. Общие замечания относительно дегидрогеназ
Структурные исследования позволили дать ясное и вполне
разумное с химической точки зрения описание стереохимических
и
каталитических условий протекания реакции переноса гидрид-
иона.
В
трех
из рассмотренных примеров присутствует кислотно-
основный
катализатор, который образует водородную связь с
карбонильной
или спиртовой группой субстрата, способствует
его правильной ориентации и стабилизирует переходное состоя-
ние
[схема (12.18)].
В
NAD
+
H
В
5
о
ч
NAD'
•+—н''Т
в
+
I
н
(12.18)
ПС
NADH