Egerton R.F. Electron Energy-Loss Spectroscopy in the Electron Microscope
Подождите немного. Документ загружается.

358 5 TEM Applications of EELS
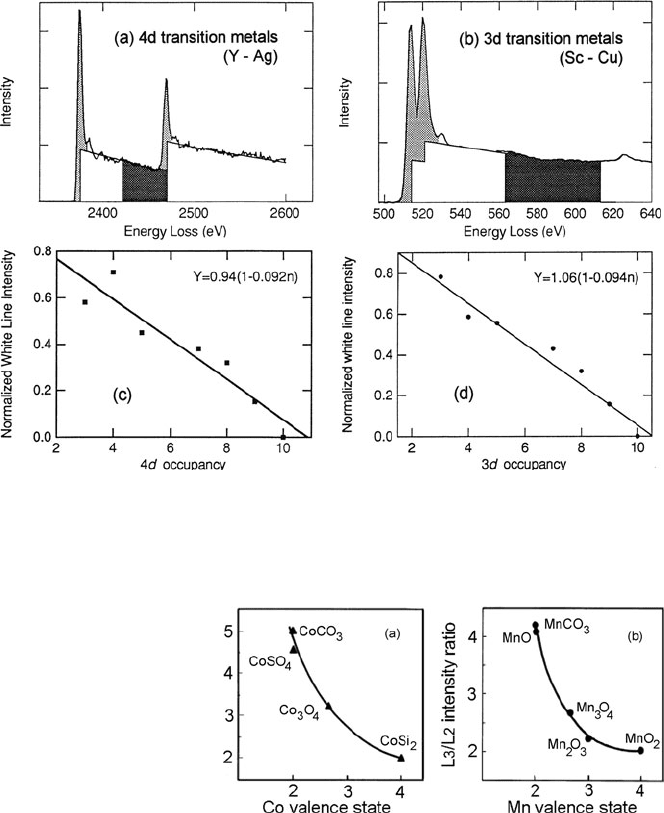
Fig. 5.41 Procedure used by Okamoto et al. (1992) to measure white-line/continuum ratio R
c
of
(a)4d transition metals and (b)3d transition metals. Graphs show the variation of R
c
with (c)4d
and (d)3d occupancy. Copyright, The Metals Society
Fig. 5.42 White-line
intensity ratio R
w
for (a)
cobalt and (b)manganese
compounds, as a function of
the cation valence. From
Wang et al. (2000), copyright
Elsevier
The white-line intensities I(L
2
) and I(L
3
) can be determined by curve fitting or by
a procedure similar to that of Fig. 5.41. The errors involved when different methods
are applied to manganese compounds are discussed by Riedl et al. (2006). Wong
(1994) has shown (for the case of nickel and its silicides) that variations in R
w
can
result from solid-state effects, besides variations in d-state occupancy.
A systematic study of a large number of chromium compounds, covering six
valences and two different spin states, was reported by Daulton and Little (2006).
Measurement of L
3
/L
2
ratio employed a method similar to Fig. 5.41, with the ratio
5.6 Structural Information from EELS 359
between the two continuum backgrounds initially set at 2 but then made equal to the
derived L
3
/L
2
ratio, this iteration being repeated 10 times. Continuum lines having
zero slope gave less scatter in the results and were consequently adopted. High-
spin Cr(II) compounds were easily distinguished by their high L
3
/L
2
ratio (1.9–2.4),
other groups being hard to distinguish on the basis of L
3
/L
2
ratio alone.
White-line ratios at the iron L
23
edge were used to study the conditions for ferro-
magnetism in amorphous alloys (Morrison et al., 1985). R
c
remains approximately
the same as germanium is added to iron, showing that the d band occupancy is unal-
tered and that the gradual loss of ferromagnetism cannot be explained in terms of
charge transfer in or out of the 3d band. However, R
w
does change, indicating redis-
tribution of electrons between the d
5/2
and d
3/2
sub-bands with a change in spin
pairing, which may account for the change in magnetic moment. Measurements on
a crystalline Cr
20
Au
80
alloy showed that the L
3
/L
2
white-line ratio increased by a
factor of 1.6 compared to pure chromium, indicating a substantial shift in spin den-
sity between j = 5/2 and j = 3/2 states, which may be the reason for a sevenfold
increase in the magnetic moment.
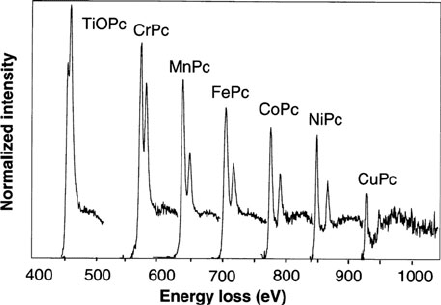
Koshino et al. (2000) recorded L
23
edges from vapor-deposited films of phthalo-
cyanine bonded to various transition metals. They found I
w
∝ 3d-state vacancy;
see Fig. 5.43. Comparison of their measured branching ratio I(L
3
)/[I(L
2
) + I(L
3
)]
with values calculated by Thole and van der Laan indicated a high spin state for the
compounds FePc, MnPc, and NiPc.
L
3
/L
2
ratio can be displayed as an intensity map. This was done using EFTEM
imaging by Wang et al. (1999) in order to display variations in the valence state of
Mn and Co in mixed-valence specimens.
5.6.4.1 Spin-State Measurements
Small changes (a few percent) in the L
3
/L
2
ratio of transition metals have been used
to determine electron spin state. For this purpose the L-edge has been measured
Fig. 5.43
Background-subtracted L
23
edges of metal
phthalocyanines. From
Koshino et al. (2000),
copyright Elsevier
360 5 TEM Applications of EELS
at selected points in the diffraction pattern, in a crystalline specimen of chosen
thickness. Differences occur because of interference between the inelastically
scattered waves and can be interpreted in terms of the mixed dynamic form fac-
tor (Schattschneider et al., 2000). Termed energy-loss magnetic chiral dichroism
(EMCD) by analogy with x-ray magnetic circular dichroism (XMCD), TEM mea-
surements offer the promise of higher spatial resolution. Applied to biomineralized
magnetite crystals in magnetotactic bacteria, a spatial r esolution of 2 nm has been
demonstrated (Stöger-Pollach et al., 2011). Zhang et al. (2009) used EMCD to con-
firm the ferromagnetic nature of ZnO nanoparticles doped with transition metal
atoms. Future aims include the study of magnetic properties at interfaces, of vital
importance for spintronic and magnetic storage devices (Schattschneider, 2011).
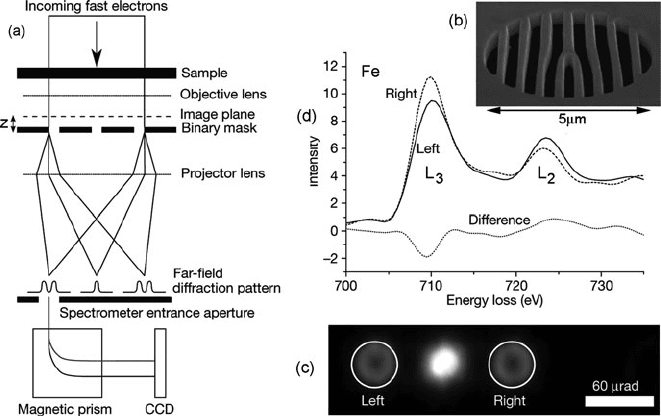
Verbeeck et al. (2010) have advocated the use of a holographic mask, taking the
form of computer-generated aperture (made by FIB milling of a Pt foil), to produce
a vortex beam in the TEM with a spiral wavefront and orbital angular momentum.
The idea was tested with a ferromagnetic iron sample; see Fig. 5.44. Compared to
the previous angle-resolved method of measuring chirality, this technique promises
improved signal/noise ratio and greater convenience, as the specimen thickness and
orientation do not need to be specially chosen.
Fig. 5.44 (a) Scheme for dichroic measurement using a computer-generated mask (b) placed a dis-
tance z beyond focus of a specimen image, together with a spectrometer entrance aperture located
(c) at the left or right sideband of the far-field diffraction pattern. ( d) L
3
and L
2
edges recorded
from a 50-nm-thick iron specimen for the two positions of the entrance aperture, together with
the difference signal that indicates asymmetry of the m =±1 dipole transitions (Verbeeck et al.,
2010). Copyright 2010, Nature Publishing Group. See also Schattschneider et al. (2008)
5.6 Structural Information from EELS 361
5.6.5 Use of Chemical Shifts
The threshold energy of an ionization edge, or changes in threshold between differ-
ent atomic environments (chemical shift), can provide information about the charge
state and atomic bonding in a solid. In the past, EELS chemical shift measure-
ments have been of limited accuracy compared to those carried out by photoelectron
spectroscopy, but the situation has improved with the development of highly sta-
ble high-voltage and spectrometer power supplies and dual-recording detectors
(Gubbens et al., 2010). As discussed in Chapter 3, the EELS chemical shift rep-
resents a net effect, involving both the initial and final states of a core–electron
transition. Coordination number also has an influence, accommodated in the concept
of coordination charge (Brydson et al., 1992b).
Muller (1999) has argued that for metals the core-loss shift arises mainly from
changes in valence band width arising from changes in atomic bonding, rather than
charge transfer. EELS could therefore provide information about the occupied states
in a metal. While the spatial difference method (Section 4.4.5) can detect core-level
shifts as small as 50 meV, these shifts could be misinterpreted as indicating a change
in the density of states at an interface (Muller, 1999).
A simple example of chemical shift is the change in energy of the π
∗
peak from
284 eV in graphite to 288 eV in calcite (Fig. 5.37) as a result of highly electroneg-
ative O atoms surrounding each C atom. Martin et al. (1989) found that the carbon
K-edge recorded from calcium alkylaryl sulfonate micelles, which contain a cal-
cium carbonate core surrounded by hydrocarbon molecules, can be represented as
a superposition of the K-edges of calcite and graphite. Peaks in the carbon K-edge
fine structure of nucleic acid bases were similarly interpreted by Isaacson (1972b)
and Johnson (1972) in terms of chemical shifts of the π
∗
peak, arising from the
different environments of carbon atoms within each molecule. They reported a peak
shift proportional to the effective charge at each site (Kunzl’s law).
For silicon alloys, Auchterlonie et al. (1989) showed that the energy of the first
peak at the Si L-edge was displaced by an amount proportional to the electronega-
tivity of the nearest-neighbor atoms (B, P, C, N, and O); see Fig. 5.45. On the basis
of this shift and the near-edge structure, their amorphous alloys could be uniquely
identified. Brydson et al. (1992a, b) explained the shape of the oxygen K-edges of
the minerals rhodizite, wollastonite, and titanite in terms of the potential at each of
the oxygen sites.
To simultaneously measure the spectra across gate-dielectric multilayers, Kimoto
et al. (1997, 1999) used spatially resolved EELS, with a slit placed in front of the
electron spectrometer. The SREELS technique ensures that high-voltage fluctua-
tions do not introduce systematic errors, as can happen when spatial resolution is
achieved by scanning a small probe.
Daulton and Little (2006) measured the chromium L
3
threshold energy of many
Cr compounds, calibrating their energy-loss spectrometer to 855.0 eV for the Ni-L
3
edge of NiO. Their results were plotted against L
3
/L
2
ratio and showed a clear cor-
relation but considerable scatter, suggesting that other factors (coordination, low- or

362 5 TEM Applications of EELS
Fig. 5.45 Energy shift of the
first peak at the silicon L
23
edge, plotted against Pauling
electronegativity (relative to
Si) of ligand atoms. From
Auchterlonie et al. (1989),
copyright Elsevier
high-spin configuration, spin–orbit interactions, Coulomb repulsion, and exchange
effects) influence the L
23
edge structure. While it appeared easier to distinguish
between the common oxidation states, Cr(III) and Cr(VI), on the basis of chemical
shift rather than L
3
/L
2
ratio, the authors argue that both measurements are neces-
sary for the unambiguous determination of valency in transition metal compounds.
Daulton et al. (2003) made similar L
3
/L
2
and chemical shift measurements on anaer-
obic Shewanella oneidensis bacteria, using an environmental cell to keep specimens
hydrated in the TEM. The data fell within the Cr(III) region (see Fig. 5.46), con-
firming these bacteria as active sites for the reduction of toxic Cr(VI) species in
chromium-contaminated water.
5.6.6 Use of Extended Fine Structure
As discussed in Section 4.6, a radial distribution function (RDF) specifying inter-
atomic distances relative to a particular element can be derived by Fourier analysis
Fig. 5.46 Chromium L
3
/L
2
ratio plotted against L
3
threshold energy for
inorganic compounds,
together with data for S.
oneidensis measured in
vacuum (square data points)
and in an environmental cell
(central data point with large
error bars). Each circular data
point represents the mean and
standard deviation of
measurements on a particular
compound. Reproduced from
Daulton et al. (2003), with
permission from Cambridge
University Press
5.6 Structural Information from EELS 363
of the extended fine structure (EXELFS), starting about 50 eV beyond an ioniza-
tion edge threshold. This procedure has been tested on various model systems and,
after correction for phase shifts, has yielded first and second nearest-neighbor dis-
tances that agree with x-ray measurements to accuracies between 0.01 and 0.001 nm
(Johnson et al., 1981b; Kambe et al., 1981; Leapman et al., 1981; Stephens and
Brown, 1981; Qian et al., 1995).
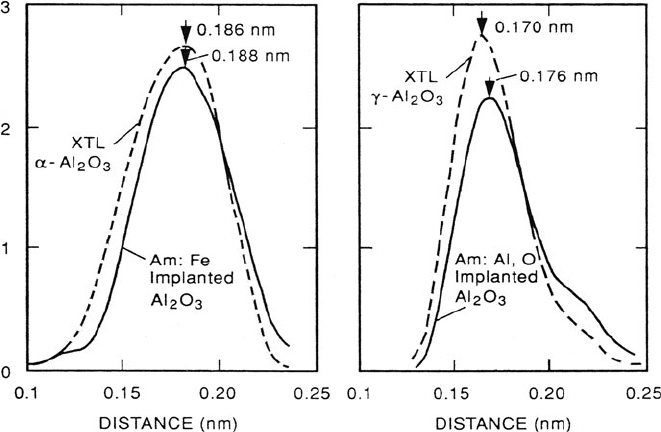
Higher precision is possible when measuring changes in interatomic distance
in specimens of similar chemical composition. The RDF of sapphire (α-Al
2
O
3
)
and amorphous (anodized) alumina in Fig. 4.22 shows a change in Al–O dis-
tance of 0.003 nm. No measurable shift in the nearest-neighbor peak occurred after
crystallizing the amorphous layer in the electron beam, consistent with the crystal-
lized material being γ - rather than α-alumina (Bourdillon et al., 1984). Aluminum
K-edge EXELFS was used to investigate the structure of ion-implanted α-Al
2
O
3
using cross-sectional TEM specimens (Sklad et al., 1992). Implantation at −185
◦
C
with 160-keV Fe ions (4 × 10
16
cm
−2
) produced a 160-nm amorphous layer that
recrystallized epitaxially to α-Al
2
O
3
upon annealing in argon at 960
◦
C. Oxygen-
edge EXELFS required a restricted energy range because of the presence of an iron
L
23
edge at 708 eV. Implantation with a stoichiometric mixture of Al and O ions
produced an amorphous layer that recrystallized into a mixture of γ -Al
2
O
3
and
epitaxial α-Al
2
O
3
, as determined from Al K-edge EXELFS; see Fig. 5.47.
Fig. 5.47 Nearest-neighbor peak in the RDF obtained from Al K-edge EXELFS of (a) α-Al
2
O
3
implanted with 160-keV Fe ions (4 × 10
16
cm
–2
), compared with the crystalline substrate and (b)
a layer implanted with a stoichiometric mixture of Al and O ions, compared with γ-Al
2
O
3
made
by annealing for 1 h in argon. Data have been corrected for phase shifts using empirical correction
factors. From Sklad et al. (1992), copyright Taylor and Francis
364 5 TEM Applications of EELS
Batson and Craven (1979) used a field-emission STEM to record K-edge
EXELFS from amorphous carbon films, revealing differences in RDF depending
on whether the substrate was mica or KCl. These results demonstrate that, even
with serial recording, EXELFS was able to provide structural information from very
small areas, below 10 nm in diameter. As always, the fundamental limit to spatial
resolution is radiation damage, but the problem can be acute in EXELFS studies
because of the need to achieve excellent signal/noise ratio in the tail of an inner-shell
edge.
Noise statistics are improved in the case of lower energy edges; the Si L-edge
from SiC was found to be sensitive to the atomic environment up to the sixth
coordination shell (Martin and Mansot, 1991). However, low-energy EXELFS is
difficult to analyze quantitatively, partly because of the high pre-edge background
and restricted energy range imposed by other ionization edges. At the opposite
extreme, EXELFS of the titanium K-edge (4966 eV), measured using 400-keV elec-
trons and parallel recording (Blanche et al., 1993), yielded results comparable with
synchrotron radiation EXAFS. Kaloyeros et al. (1988) used boron edge EXELFS to
investigate the high-temperature stability of amorphous films of titanium diboride,
made by electron-beam evaporation onto liquid-nitrogen-cooled substrates.
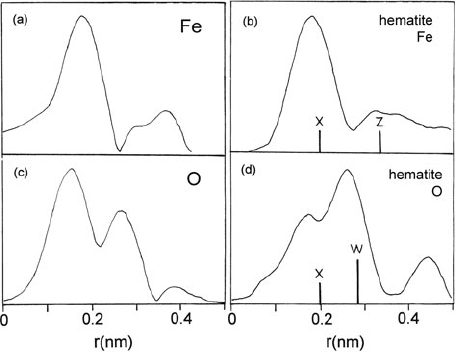
As an example of a biological application, Fig. 5.48 shows RDFs recorded from
iron-rich clusters (siderosomes) extracted from lung fluids of a patient suffering
from silicosis (Diociaiuti et al., 1995). The Fe-L
23
RDF is similar to that recorded
from a hematite standard but the RDF from the oxygen K-edge exhibits a displaced
Fig. 5.48 Radial distribution functions for (a)Featomsand(c) O atoms, obtained from EXELFS
of alveolar macrophages (siderosomes). The iron and oxygen RDF recorded from hematite are
givenin(b)and(d), where vertical bars marked X, Z, and W show the expected Fe–O, Fe–Fe, and
O–O interatomic distances. From Diociaiuti et al. (1995), copyright EDP Sciences (Les Éditions
de Physique)
5.6 Structural Information from EELS 365
nearest-neighbor peak and a second-nearest-neighbor (O–O) peak of reduced inten-
sity compared to hematite. These discrepancies were explained by assuming that
oxygen is also present in a protein coat surrounding the biomineral core, forming
short O=C bonds that shift the center of the first peak to lower radius. Diociaiuti
and colleagues (1991, 1992a, b) recorded EXELFS from chromium, copper, and
palladium clusters in order to quantify the increase in nearest-neighbor distance (up
to 5%) with decreasing particle size and the effect of oxidation.
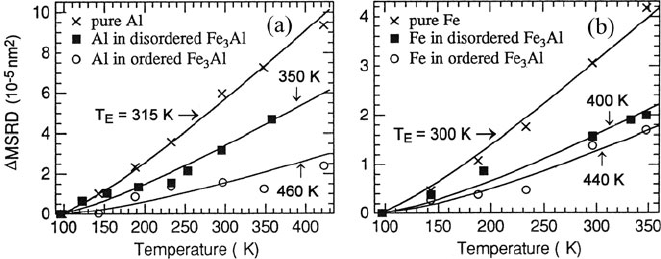
EXELFS measurements as a function of temperature enabled Okamoto et al.
(1992) to study ordering in undercooled alloys, particularly chemical short-range
order that is difficult to measure by techniques such as diffuse x-ray scattering. The
temperature dependence of the mean-square relative displacement (MSRD) was
used to deduce Einstein and Debye temperatures, good agreement being obtained
with force-constant theory and with previous experimental data. Differences in
Einstein temperature between ordered and disordered alloys, reflecting difference
in vibrational states, are illustrated in Fig. 5.49. Since the EXELFS data can in prin-
ciple be obtained from small specimen volumes, measurement of a local Debye
temperature might usefully characterize the defect density at interfaces or in small
precipitates (Disko et al., 1989).
To achieve the 0.1% statistical accuracy that is desirable for analyzing EXELFS
data, as many as 10
6
electrons need to be recorded within each resolution element
(typically 2 eV). Careful monitoring of relative peak heights within each readout
can alert the operator to damage and changes in specimen thickness within the beam
(Qian et al., 1995). High-voltage and spectrometer drift can be corrected by shifting
successive readouts back into register. It is important to carefully remove diode array
dark current and gain variations (Section 2.5.5).
Even with parallel recording, EXELFS analysis requires an incident electron
exposure of typically 10
−7
C. For a 1-μm-diameter incident beam, this is equivalent
Fig. 5.49 Temperature dependence of the nearest-neighbor mean-square relative displacement
(MSRD) measured for (a)Aland(b) Fe a toms in pure elements and in chemically disordered
and ordered Fe
3
Al. All values are relative to measurements made at 97 K; solid lines represent
the Einstein model, with an Einstein temperature T
E
as indicated. From Okamoto et al. (1992),
copyright TMS Publications
366 5 TEM Applications of EELS
to 1 C/cm
2
dose, enough to destroy the structure of most organic specimens and even
some inorganic ones (see Section 5.7.3). Because most damage-producing inelastic
collisions involve energy losses outside the EXELFS region, electrons may produce
more damage than the monochromatic x-rays used in EXAFS studies (Isaacson and
Utlaut, 1978; Hitchcock et al., 2008). If there exist materials in which structural
damage takes place only as a result of inner-shell scattering, this conclusion would
not apply (Stern, 1982). Provided radiation damage is not a problem, the TEM is
competitive with synchrotron sources in the sense that the EXELFS recording time
is typically less than that of EXAFS for ionization edges below 3 keV (Isaacson and
Utlaut, 1978;Stern,1982).
The core-loss intensity is improved by increasing the collection semi-angle β,
but at the expense of a higher pre-edge background (Section 3.5). If β is too large,
nondipole contributions complicate the EXELFS analysis, but such effects are usu-
ally assumed to be small (Leapman et al., 1981;Disko,1981); see Section 3.8.2.For
100-keV electrons, a semi-angle in the range 10–20 mrad should allow dipole theory
to be used, while transmitting typically half of the core-loss scattering. However, this
large angle will average out the directional dependence of EXELFS (Section 3.9),
so a much smaller value of β is necessary to study the directionality of bonding.
5.6.7 Electron–Compton (ECOSS) Measurements
Electron energy-loss spectra are most often recorded with a collection aperture cen-
tered on the optic axis, around the unscattered beam. If this aperture is displaced or
the incident beam tilted through a few degrees so that only large-angle scattering is
collected, a new spectral feature emerges at high energy loss in the form of a broad
peak; see Fig. 5.50. Known as an electron-Compton profile, this peak represents a
cross section (at constant scattering vector q) through the Bethe ridge; see Figs. 3.31
and 3.36. Its center corresponds to an energy loss E that satisfies Eq. (3.132),the
scattering angle θ
r
being determined by the collection aperture displacement or the
tilt of the incident beam. For sin θ
r
<< 1 and E << E
0
, Eq. (3.132) is equiv-
alent to the relation E = γ
2
Tθ
2
r
for Rutherford scattering from a free stationary
electron.
The fact that the atomic electrons are not free is indicated by the width of the
Compton profile, which is a measure of the electron binding energy. Therefore,
although the peak contains overlapping contributions from both outer- and inner-
shell electrons, the latter give a much broader energy distribution and contribute
mainly to the tails of the profile. Conversely, the central region represents mainly
scattering from bonding (valence) electrons.
The spread of the Compton peak can also be thought of as a Doppler broaden-
ing due to the “orbital” velocity or momentum distribution of the atomic electrons,
closely related to the electron wavefunctions. Quantitative interpretation (Williams
et al., 1981) is rather similar to the Fourier method of EXELFS analysis. After sub-
tracting t he background contribution from the tails of lower energy processes, the

5.6 Structural Information from EELS 367
Fig. 5.50 (a) Energy-loss spectrum of amorphous carbon, recorded using 120-keV electrons scat-
tered through an angle of 100 mrad. Zero-loss and plasmon peaks are visible (because of elastic
and multiple scattering), in addition to the carbon L-edge and Compton peak. The dashed curve
shows a computed free-atom Compton profile. (b) Reciprocal form factor derived for amorphous
carbon (solid line) and for two basal plane directions in graphite. From Williams and Bourdillon
(1982), copyright IOP Publishing
energy scale is converted to one of momentum (or wave number k) of the atomic
electron. The Fourier transform of the resulting profile (Fig. 5.50b) is known as a
reciprocal form factor B(r) and is the autocorrelation function of the ground-state
atomic wavefunction, in a direction specified by the scattering vector q (i.e., by
the azimuthal location of the detector aperture). If the specimen is an insulating or
a semiconducting crystal, zero crossings in B(r) are expected to coincide with the
lattice s pacings.
The optimum scattering angle for recording the electron-Compton profile from
a light element sample (such as graphite) appears to be about 100 mrad for
60–100 keV electrons, resulting in a profile whose center lies at about 1 keV loss.
An energy resolution of 10 eV is sufficient, but should be combined with an angu-
lar resolution of about 3 mrad (Williams et al., 1984). The signal/background ratio
at the Compton peak is maximized by making the sample as thin as possible,
indicating that the background arises from plural or multiple scattering, chiefly
large-angle elastic (or phonon) scattering accompanied by one or more small-
angle (low-loss) inelastic events. Because of the large width of the Compton peak,
accurate subtraction of the background is not easy. To obtain a tolerably low back-
ground, the specimen thickness should be less than about 30 nm in the case of
100 keV incident energy and a low-Z element such as carbon. Even thinner sam-
ples are appropriate for higher Z elements and would result in l onger recording
times.
Because of the need for both angular and energy discrimination, the collec-
tion efficiency of the Compton signal is of the order of 10
−6
, assuming parallel
recording. Adequate statistics within the Compton peak involve recording at least
10
6
electrons, requiring an incident exposure ≈3 × 10
−3
C, equivalent to a dose of