Caballero B. (ed.) Encyclopaedia of Food Science, Food Technology and Nutrition. Ten-Volume Set
Подождите немного. Документ загружается.


Spectroscopy: Nuclear Magnetic Resonance; Visible
Spectroscopy and Colorimetry; Tocopherols: Properties
and Determination
Further Reading
Brumley WC, Sheppard AJ, Rudolf TS et al. (1985) Mass
spectrometry and identification of sterols in vegetable
oils as butyryl esters and relative quantitation by gas
chromatography with flame ionization detection. Jour-
nal of the Association of Official Analytical Chemists
68: 701–709.
Christie WW (1987) High-performance Liquid Chroma-
tography and Lipids: A Practical Guide, 1st edn. New
York: Pergamon Press.
Faulkner WR and Meites S (eds) (1982) Selected Methods
for the Small Clinical Chemistry Laboratory, vol. 9, pp.
157–183. Washington, DC: American Association of
Clinical Chemistry.
Fieser LF and Fieser M (1949) Natural Products Related to
Phenanthrene, 3rd edn. New York: Reinhold.
Macrae R (1988) HPLC in Food Analysis, 2nd edn.
London: Academic Press.
Sheppard AJ, Newkirk DR, Hubbard WD and Osgood T
(1977) Gas–liquid chromatographic determination
of cholesterol and other sterols in foods. Journal of
the Association of Official Analytical Chemists 60:
1302–1306.
Windaus A (1932) Uber die Konstitution des Cholesterins
und der Gallensauren. Zeitschrift fu
¨
r Physiologische
Chemie 213: 147–187.
Yasaei P, Sheppard AJ, Brumley WC, Mazzola EP and
Aldridge MH (1989) Structural proof of cholesterol isol-
ated from plants. II. Identification and verification by
13
C nuclear magnetic resonance spectroscopy and mass
spectrometry. Journal of Micronutrient Analysis 5:
259–267.
Yasaei P, Sheppard AJ, Rudolf TS, Shen C-SJ and Aldridge
MH (1989) Structural proof of cholesterol isolated from
plants. I. Isolation and preliminary identification by
chromatography and infrared spectroscopy. Journal of
Micronutrient Analysis 5: 238–245.
Absorption, Function, and
Metabolism
D R Arnold and P O Kwiterovich Jr, Johns Hopkins
University, Baltimore, MD, USA
Copyright 2003, Elsevier Science Ltd. All Rights Reserved.
Cholesterol Metabolism
0001 Cholesterol metabolism in humans is complex. Chol-
esterol is either supplied from the diet (exogenous)
or synthesized de novo by many cells of the body
(endogenous). The major factors in the diet that may
increase the blood cholesterol level are high intakes of
cholesterol itself, or of saturated fats and excessive
calories. The liver is one of the major sites of en-
dogenous cholesterol synthesis. The pool of choles-
terol in the liver is tightly regulated and reflects the
input of cholesterol from the diet, the biosynthesis of
cholesterol, the secretion and uptake of cholesterol
from plasma lipoproteins, the conversion of choles-
terol into bile, and the reuptake of biliary cholesterol
and bile acids from the intestine to the liver. The
metabolic exogenous and endogenous pathways of
cholesterol metabolism are described in this chapter.
The concentration of cholesterol in the blood in
fasting normal humans is the result of the metabolism
of cholesterol from exogenous and endogenous
sources. Environmental factors such as dietary fatty
acids, and metabolic perturbations such as diabetes
and obesity as well as genetic factors also influence
the level of cholesterol in blood. Consequently, after
several decades, hypercholesterolemia causes athero-
sclerotic vascular disease. Dieting, exercise, and sev-
eral drugs are available as tools to lower blood
cholesterol levels, and the mechanisms by which
these interventions lower elevated cholesterol levels
are discussed here.
Cholesterol – Chemical Structure and
Importance
0002Cholesterol (cholest-5-en-3-ol (3-beta) cholesterol)
and cholesterol metabolites and esters are major com-
ponents of the plasma membrane and of many other
cellular organelles in animals. On average, 1.5 g of
cholesterol is present per kilogram of body weight;
some tissues like fat, brain, and liver contain higher
amounts of cholesterol (e.g., liver contains 4.5 g
kg
1
). Cholesterol is a water-insoluble lipid, made
of a so-called sterol backbone with one hydroxy
group and one double bond, together with a side-
chain made up of eight carbon atoms. Cholesterol is
chemically distinctly different from two other lipids,
triacylglycerols and phopholipids. The steroid back-
bone of cholesterol also constitutes the fundamental
structure of bile acids (with cholic acid and cheno-
deoxycholic acid being the primary bile acids), certain
fat-soluble vitamins, such as vitamin D
3
, steroid hor-
mones (cortisone and aldosterone), and sex hormones
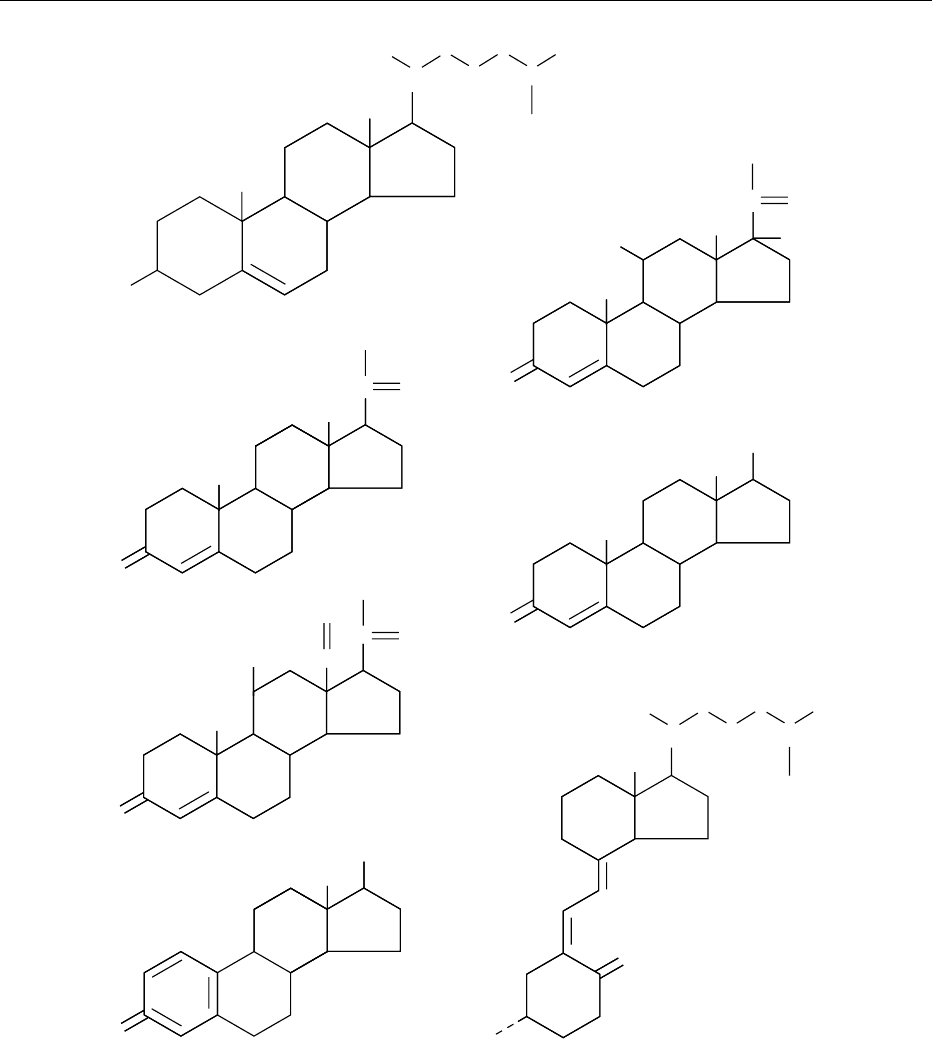
(estradiol and testosterone, Figure 1).
0003Cholesterol is primarily transported through blood
on low-density lipoproteins (LDL). When present in
elevated amounts, LDL cholesterol causes ‘hardening
of the arteries’ or atherosclerotic cardiovascular dis-
ease (ASCVD). The evidence for a causal relationship
1226 CHOLESTEROL/Absorption, Function, and Metabolism
between elevated blood LDL cholesterol and ASCVD
comes from many research studies in various disci-
plines (feeding studies of animals, epidemiological
surveys, studies of genetic abnormalities of lipid dis-
orders, and large, placebo-controlled, double-blind
clinical trials).
History
0004Poulletier de la Salle discovered cholesterol in gall-
stones in 1769, but Chevreul gave it the name ‘cho-
lesterine’ in 1815. Ignatowsky first demonstrated in
1908 the connection between intake of cholesterol in
OH
OH
HO
HO
OH
CH
3
CH
3
CH
3
CH
3
CH
3
CH
2
OH
CH
2
OH
CH
3
CH
3
CH
CH
2
CH
3
CH
3
CH
3
CH
3
CH
3
CH
CH
CH
2
CH
3
CH
CH
3
CH
3
CH
2
CH
2
CH
3
CH
3
CH
3
CH
2
CH
2
CH
2
O
O
HO
O
O
HC
C
C
C
O
O
O
O
OH
OH
Progesterone
Cholesterol
Aldosterone
Estradiol
Cortisol
Testosterone
Vitamin D
3
fig0001 Figure 1 Biochemical structure of cholesterol in comparison with other hormones derived from cholesterol. Cholesterol is
homologous to several steroid hormones: cortisol, a stress hormone, progesterone, testosterone and estradiol (sex hormones),
aldosterone (important for blood pressure control and salt–fluid balance) and vitamin D
3
(important for calcium absorption in the gut
and homeostasis).
CHOLESTEROL/Absorption, Function, and Metabolism 1227
the diet of rabbits and the development of arterio-
sclerosis. Anitschkow demonstrated in 1913 that it
was cholesterol feeding only that caused athero-
sclerotic changes in the blood vessels of rabbits.
Cholesterol De-novo Production
0005 Cholesterol can be either absorbed from food or
made de novo in multiple human tissues in varying
amounts. The biosynthesis of cholesterol starts with
acetyl coenzyme A (acetyl CoA). Three molecules of
acetyl CoA are condensed to form 3-hydroxy-3-
methylglutaryl CoA (HMG CoA). The conversion of
HMG CoA to mevalonate by HMG CoA reductase is
the rate-limiting step of cholesterol synthesis. A
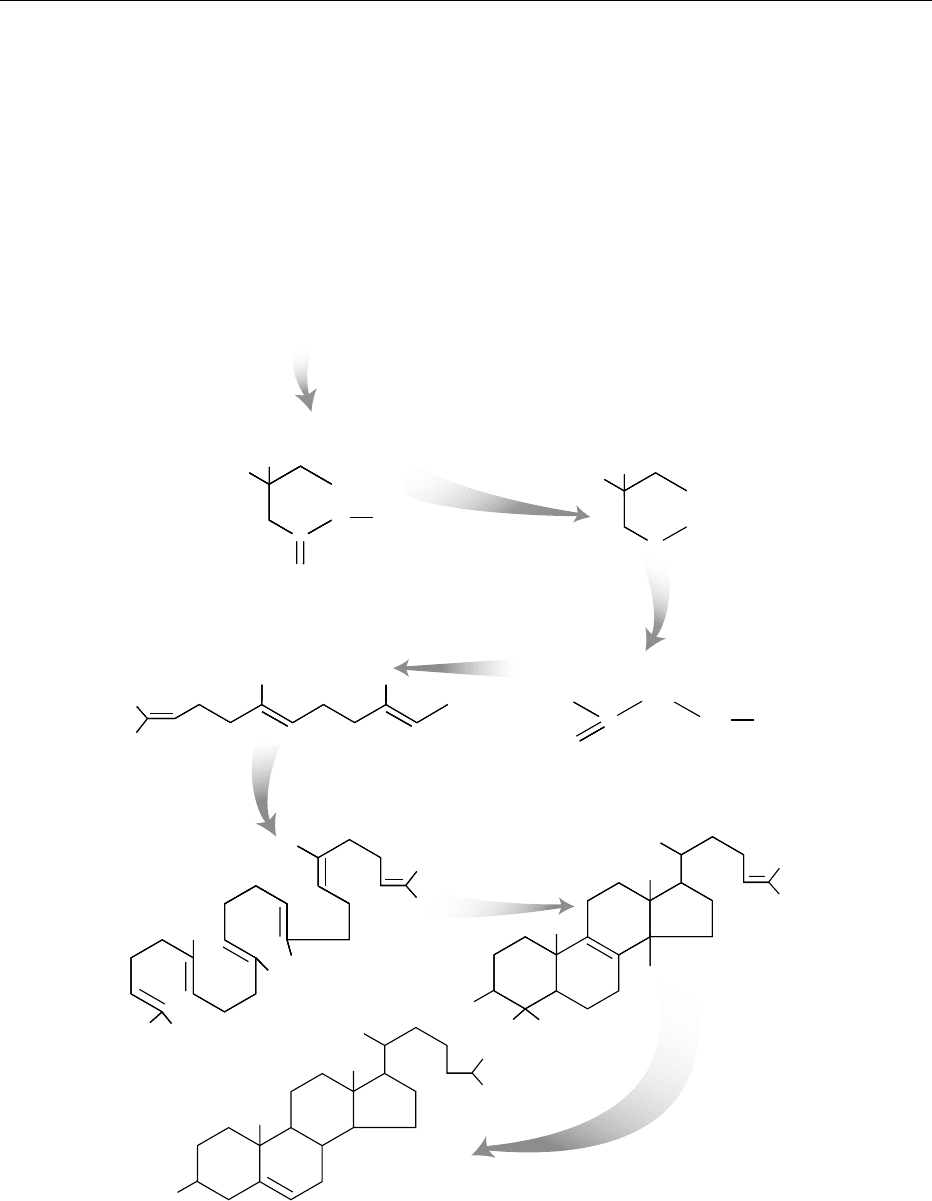
schematic diagram summarizing the de-novo biosyn-
thesis of cholesterol is shown in Figure 2. While most
of the sterol pool of the body is made de novo in
extrahepatic tissues, the liver is a major organ
involved both in the synthesis of cholesterol and
COOH
S
C
O
HO
3-Hydroxy-3-methylglutaryl CoA
(HMG CoA)
Acetoacetyl CoA + acetyl CoA
Farnesyl pyrophosphate (FP)
OPP
Squalene
HO
Cholesterol
Mevalonic acid
HO
COOH
OH
C
HMG CoA
reductase
3 - Isopentenyl pyrophosphate (IPP)
H
3
C
H
2
C
CH
2
CH
2
C
OPP
Lanosterol
c. 20
Enzymatic
steps
HO
CoA
fig0002 Figure 2 De-novo biosynthesis of cholesterol. The biosynthesis of cholesterol begins with the condensation of three acetyl
molecules to form 3-hydroxy-3-methylglutaryl CoA (HMG CoA). The acetyl-CoA can be derived from glucose, fatty acids, or amino
acids. Reduction of HMG CoA produces mevalonate, which gets further processed to isopentenyl pyrophosphate (IPP). Three
molecules of IPP are condensed to make up farnesyl pyrophosphate (FP). Two molecules of FP are used to make up squalene,
which gets converted by three enzymatic steps to lanosterol. Approximately 20 additional chemical steps are needed to make up
cholesterol.
1228 CHOLESTEROL/Absorption, Function, and Metabolism

in the processing and repackaging of cholesterol and
other lipids absorbed from the diet. By using H
3
-
labeled water, the average production of cholesterol
in man has been estimated to about 9 mg per kilo-
gram per day. Using this technique, the most active
cholesterol processing tissue is the liver followed by
the gastrointestinal tract, skin, striated muscle and
bone marrow while minor amounts are made in the
adrenal glands, and other endocrine organs like ovar-
ies and testis.
Cholesterol from Diet and Factors
Influencing Cholesterol Absorption/
de novo
Synthesis
0006 The amount of synthesis of cholesterol in mammalian
liver is tightly controlled by multiple factors. Modu-
lating dietary factors that affect plasma cholesterol
levels include energy restriction in the form of de-
creased lipid and carbohydrate intake (diet), meal
frequency, dietary fat type (saturated, unsaturated or
trans), and cholesterol content of the food. Plants
make sitosterol but not cholesterol, and an average
American diet contains about 100–200 mg of this
plant sterol per day. In the intestine, inhibition of
the uptake of cholesterol occurs with intake of these
so-called phytosterols, and this can lower the blood
cholesterol level. Diseases of the liver, or exposure to
drugs and toxins such as alcohol, can easily disturb
the ability of the liver to handle cholesterol and other
fats, and may result in the initiation of ‘fatty degener-
ation,’ thus demonstrating further the importance of
the liver in the metabolic processing of lipids.
0007 Cholesterol in the intestinal lumen comes either
from the diet (250–500 mg) or the bile (c. 600–
1000 mg). While bile acids, such as cholic acid and
chendeoxycholic acid, are nearly completely recircu-
lated, only about 50% of the dietary cholesterol is
absorbed at an average intake of 300–600 mg per day.
Formulas developed by Keys and by Hegstedt and
coworkers predict the average increase in the blood
cholesterol level that will result from a given change
in the amounts of dietary cholesterol, saturated and
unsaturated fatty acids. The Hegstedt formula is as
follows:
< Blood cholesterol ðmg dl
1
Þ
¼ 2:16 < S 1:65 < P þ 0:068 < C,
where < symbolizes a change in; S ¼ dietary saturated
fatty acids (% of total calories), P ¼ dietary poly-
unsaturated fatty acids (% total calories) and C ¼
dietary cholesterol (mg per day). Examples of
polyunsaturated fats include linoleic acid (C18:2),
linolenic acid (C18:3), and arachidonic acid
(C20:4). Linoleic acid is the essential fatty acid,
which can be elongated and desaturated to form
other polyunsaturated fats. Fish oils include eicosa-
pentaenoic acid (EPA: C20:5) and docosahexaenoic
acid (DHA: C22:6).
0008Without drug treatment, dietary cholesterol and
saturated fats, and the de novo production and re-
moval of LDL, determine the blood LDL-cholesterol
level. Saturated fatty acids, particularly with 14 and
16 carbons, suppress the receptor-mediated uptake of
LDL and increase the LDL cholesterol level, whereas
unsaturated fatty acids like 9-cis 18:1 increase
receptor-mediated LDL uptake and lower the LDL
cholesterol level.
Excretion of Cholesterol
0009The human does not have an enzyme that can degrade
the sterol ring of cholesterol; the backbone/sterol ring
of cholesterol thus has to be excreted in the bile and
stool. All cholesterol metabolites, including hor-
mones and vitamin metabolites, are excreted as bile
acids or metabolites in the intestine, but they are
partially reabsorbed in the intestine via the entero-
hepatic pathway. The sterol ring of hormones and
vitamins are metabolized (hydroxylated/sulfonu-
rated) by multiple enzymes in the liver to make the
sterol more water-soluble, which promotes excretion
in bile and the urine as well. The delicate balance of
intake and excretion of cholesterol and cholesterol
products is tightly regulated. Evolutionarily, excess
cholesterol must be avoided inside cells as it can be
toxic. Excess cholesterol in the bloodstream is also
undesirable, as it causes hardening of the arteries
(atherosclerosis) and consequently, ASCVD.
Lipoproteins
0010To make lipids soluble in blood, they usually are
packaged inside a lipid-rich core, around which
are wrapped amphipathic proteins, whose nonpolar
amino acid groups are exposed to the lipid core,
whereas their polar or charged amino acids are
oriented to the outer surface, making the entire lipo-
protein particle water-soluble. The proteins associ-
ated with lipoproteins are called apolipoproteins
and act as surface components of the particle, as
enzymes key to lipid metabolism in blood, or as
ligands to cell-surface receptors promoting the bind-
ing and uptake of lipoproteins. A list of these apo-
lipoproteins explaining their functions are listed in
Table 1. Multiple apolipoproteins can be present on
the surface of the lipoprotein particle and provide
information about the tissue of origin and the func-
tion of the lipoprotein. For example, apoB-48, a
CHOLESTEROL/Absorption, Function, and Metabolism 1229

truncated version of apolipoprotein B, is only made in
the intestine and is present only on chylomicrons and
chylomicron remnants, whereas apoB-100 is made in
the liver and is the major apolipoprotein on very-low-
density lipoproteins(VLDL) andtheproducts ofVLDL
catabolism, VLDL remnants and intermediate density
lipoproteins (IDL), and LDL. Multiple enzymes are
present on the capillary walls and on the lipoprotein
particles that metabolize/transfer the lipids present in
the particles (see Table 2 and also below).
0011 There are principally five major classes of lipopro-
tein particles in the blood: chylomicrons (CM), VLDL,
IDL, LDL, and high-density lipoproteins (HDL),
based on their physicochemical properties. This clas-
sification is based on a separation via a salt density
gradient (chylomicrons are the lightest particles,
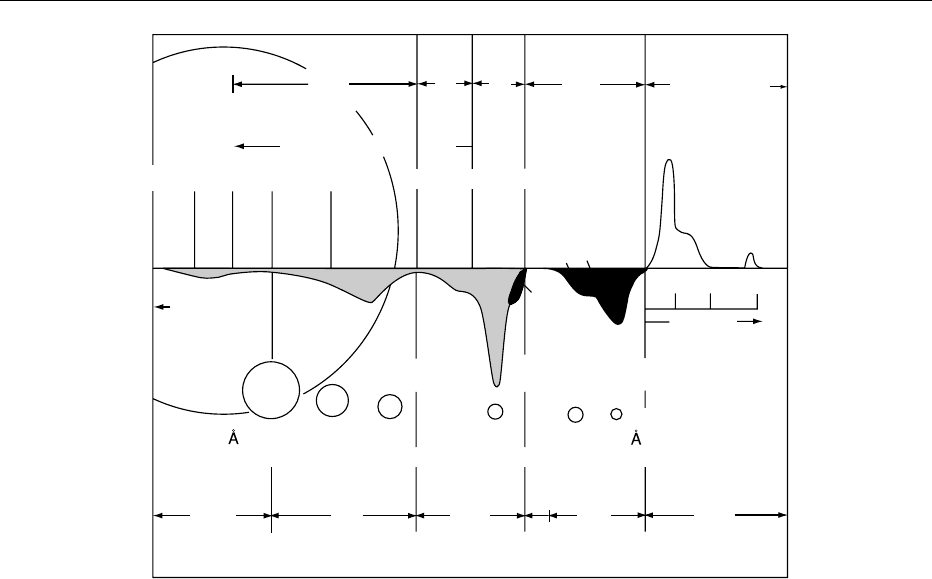
whereas HDL particles are the heaviest; see Figure 3
for further explanations).
Chylomicrons and Transport of Dietary (Exogenous)
Fat
0012 Dietary cholesterol and triacylglycerol are emulsified
in the intestinal lumen by bile salts; the cholesteryl
esters are hydrolyzed into unesterified cholesterol
and free fatty acids (FFA), and the triacylglycerol
into 2-monoglycerol and FFA. These constituents
are absorbed into the intestinal epithelial cells where
they are repackaged into chylomicrons. Chylo-
microns are large (c.1mm) and their lipid-containing
core is surrounded on the surface by apoproteins
which are amphipathic molecules that allow the
lipid core to be soluble in aqueous lymph fluid and
blood. These particles contain the apolipoproteins B-
48, A-1, A-2, and A-4. The secretion of the chylomi-
crons from the intestinal cells into the lymph requires
the presence of apoB-48. The lymph from the gut
collects in the thoracic duct and enters the blood
stream via the right subclavian vein. Even before
the chylomicrons reach the systemic circulation, the
apoA-1 content is decreased, and apoE, apoC-1,
apoC-2, and apoC-3 are acquired as a result of
HDL particles interacting with chylomicrons in the
intestinal lymph capillaries.
0013Chylomicrons then interact with endothelial cells
in the arterioles lining a number of tissues. The tri-
acylglycerols in the core of the chylomicrons, through
the interaction of apoC-2 with the enzyme lipopro-
tein lipase (LPL) bound to the surface of endothelial
tbl0001 Table 1 Characteristics of apolipoproteins: their origins, functions and distribution in plasma lipoprotein classes
Apolipoprotein Origin Function Presentin lipoprotein
classes
Molecular
weight (kDa)
A-1 Liver, intestine Activator of LCAT CM, HDL 29 016
A-2 Liver, intestine Inhibits HL and VLDL hydrolysis CM, HDL 17 414
A-4 Liver, intestine Activates LCAT CM, HDL 44 465
B B100 liver Ligand for LDL receptor B100!VLDL, LDL 512 723
B48 intestine Necessary for secretion of intestinal and hepatic fat B48!CM 241 000
C-1 Liver Cofactor for LCAT CM, VLDL, HDL 6 630
C-2 Liver Activates LPL, cofactor for LCAT CM, VLDL, HDL 8 900
C-3 Liver, intestine Inhibits LPL/inhibits binding of ApoE to the LDL
receptor
CM, VLDL, HDL 8 800
D Many sources Reverse cholesterol transport HDL 19 000
E (2,3, and 4) Liver, peripheral tissues Ligand for LDL receptor and LRP receptor CM, VLDL, HDL 34 145
CM, chylomicron; LDL, low-density lipoprotein; HDL, high-density lipoprotein; VLDL, very-low-density lipoprotein; LPL, lipoprotein lipase; HL, hepatic
lipase; LCAT, lecithin cholesterol acyltransferase; LRP, low-density lipoprotein receptor-like protein.
tbl0002 Table 2 Characteristics of key enzymes and transfer proteins of lipoprotein particles in the blood
Enzyme Origin Function Lipoproteinclasses
involved
Molecular
weight (kDa)
LPL Capillaries of muscle and
adipose tissue
Hydrolyzes triglycerides and phospholipids CM, large VLDL 50 394
HL Liver Hydrolyzes triglycerides and phospholipids Small VLDL, IDL, HDL 53 222
LCAT Liver Converts free cholesterol from cell membranes to
cholesterol esters
HDL 47 090
CETP Liver, spleen, adipose tissue Transfers cholesteryl esters from HDL to ApoB
containing lipoproteins
HDL, LDL, VLDL 74 000
PTP Pancreas, adipose tissue, lung Transfers the majority of phospholipids in plasma HDL 81 000
CM , chylomicron; LDL , low-density lipoprotein; HDL , high-density lipoprotein; VLDL , very-low-density lipoprotein; LPL , lipoprotein lipase; HL , hepatic
lipase; LCAT, lecithin cholesterol acyltransferase; CETP, cholesterol transfer protein; PTP, phospholipid transfer protein.
1230 CHOLESTEROL/Absorption, Function, and Metabolism
cells, are hydrolyzed into FFA and glycerol, which are
then absorbed into adipocytes and other cells. During
hydrolysis, phospholipids and apolipoproteins are
removed from the surface of the chylomicrons and
transferred to HDL. By removing apoC-2, an activa-
tor of LPL, the process starts to slow down. The
resultant chylomicron remnant, which contains less
triacylglycerols and relatively more esterified choles-
terol, is removed rapidly from the circulation by its
interaction with the chylomicron receptor (low-
density, lipoprotein receptor-like protein (LRP)) on
the surface of the liver cells. The LDL receptor and
proteoglycans can also facilitate the uptake of chylo-
micron remnants into the liver. ApoE is the ligand
that permits the binding and uptake of the chylomi-
cron remnant.
Very-low-density Lipoproteins and Transport of
Hepatic (Endogenous) Fat
0014 In the liver, cholesterol, triacylglycerols, and other
fats are packaged into the VLDL particle that is
surrounded by apoB-100. Triacylglycerol and choles-
teryl ester production is necessary for VLDL synthesis
by the liver. The availability of apoB-100 affects both
the assembly and secretion of the VLDL. The apoB-
100 gene is constitutively expressed.
0015The amount of apoB-100 available is regulated by
the rate of proteolysis of apoB-100. When excess lipid
is available, proteolysis is decreased, and more apoB-
100 is available for VLDL production. When lipid is
depleted, apoB-100 proteolysis increases. The triacyl-
glycerols and apoB-100 interaction requires the
presence of the microsomal triacylglycerol transfer
protein (MTP). MTP is a heteromer that consists of
two subunits, a protein widely distributed in many
cells, called disulfide isomerase, and a unique 97-kDa
large subunit. A mutation in the 97-kDa protein pre-
vents the association of lipid with apoB-100, leading
to little, if any, secretion of VLDL and no production
of IDL and LDL. At least a dozen mutations in the
97-kDa protein have been described in patients with
the recessive disorder, abetalipoproteinemia.
VLDL IDL LDL HDL
UC residue
10
5
10
4
10
3
400 100 20 12 0
510 20
Flotation rate
(s
f
)
Chylomicra
500
78
700 400 300 200 100
Molecular diameter
(Assumed spherical)
0−50
(0−2)
120
(20)
300
(135)
250
(45)
6000
(0)
1.007
g ml
−1
1.063
g ml
−1
1.200
g ml
−1
(pre-β) (pre-β) (β) (α)
S rate
1
2
3
Average concentration (Chol, mg dl
−1
)
fig0003 Figure 3 Characteristics of the major lipoproteins present in normal human plasma. Flotation rates are measured in S
f
units. The
flotation of VLDL, IDL and LDL is performed at a salt gradient of 1.063 g ml
1
, whereas a density of 1.200 g ml
1
is used for HDL. During
ultracentrifugation at 200 000 g for 1 h, a light beam is directed through an aperture in the cell and the changes in diffraction recorded to
outline the boundaries caused by the floating lipoproteins. The class of lipoprotein is indicated at the top, corresponding to
abbreviations and a description given in the text. ‘UC-residue’ describes nonlipid plasma proteins that sediment at a density of
1.200 g ml
1
. The amount of each lipoprotein class is proportional to the area under each curve shown numerically at the bottom. There
are three subclasses of HDL (1, 2, and 3), and these are indicated by numbers and colored black. HDL1 occurs only in hyperlipemic
plasma. The approximate diameter of each lipoprotein class particle is shown in A
˚
ngstro
¨
ms (1 A
˚
¼ 0.1 nm), and the average amount of
plasma cholesterol present in each class of lipoprotein is shown in parentheses (for example, 45 mg dl
1
for HDL lipoproteins). From
Olson RE (1998) Discovery of the lipoproteins: their role in fat transport and their significance as risk factors. Journal of Nutrition
128(Supplement 2): 439S–443S, with permission.
CHOLESTEROL/Absorption, Function, and Metabolism 1231

0016 VLDL is secreted from the liver into the blood-
stream surrounded by apoB-100, apoE, and the
apoCs. Once in the bloodstream, the apoC-2 on
VLDL binds to the same LPL site as chylomicrons in
the capillaries surrounding various tissues.
Intermediate-density lipoproteins
0017 As the core triacylglycerol in VLDL is hydrolyzed, a
remnant VLDL is formed, and the released FFA are
taken up by tissues. Upon further hydrolysis of tri-
acylglycerol, the final remnant, IDL, is formed. The
liver takes up some IDL through the interaction of
apoE with the LDL (B, E) receptor. The rest of the
IDL is modified at the surface of the liver by hepatic
lipase (HL), producing the cholesteryl ester-enriched
LDL.
Low-density Lipoproteins – Production, Uptake,
and Catabolism
0018 The majority of LDL is formed from VLDL. The
major apolipoprotein of LDL is apoB-100, which
serves as the ligand for the binding of LDL to the
LDL (B, E) receptor in clustered pits on the surface
of liver and peripheral cells. The bound LDL and its
receptor are internalized into the cell. LDL are taken
up and degraded in the lysosomes, while the LDL
receptor is released and recycled back to the cell
surface. Cholesteryl esters in the core of LDL are
hydrolyzed into unesterified (free) cholesterol and
FFA. The enzyme acyl cholesterol acyl transferase
(ACAT) controls the cellular levels of unesterified
(free) cholesterol versus cholesteryl esters. Monoun-
saturated fat such as oleic acid is a preferred substrate
for ACAT, whereas saturated FFA is not. In the pres-
ence of excess dietary saturated FFA, less cholesterol
is esterified, and the excess cholesterol inhibits the
release of the transcription factor from the nuclear
membrane. Thus, less of this factor is available to go
into the nucleus where it ordinarily upregulates the
transcription of both the LDL receptor gene and the
gene for HMG CoA reductase, the rate-limiting
enzyme of cholesterol production (see below).
High-density Lipoproteins and Reverse Cholesterol
Transport
0019 HDL play a paramount role in the reverse cholesterol
transport pathway, which mediates the removal of
cholesterol from peripheral cells for transport back
to the liver. HDL are secreted from the liver and
intestine as nascent, disc-shaped particles that consist
primarily of phospholipids and apoA-1, with very
little cholesterol. The nascent HDL interacts with
the ABCA1 transport protein on the surface of
peripheral cells such as macrophages and removes
unesterified cholesterol. In the bloodstream, the free
cholesterol is esterified by lecithin cholesterol acyl
transferase (LCAT) and its cofactor, apoA-1, produ-
cing a spherical HDL particle with a cholesteryl ester
in its core. The core cholesteryl ester in HDL can be
selectively taken up in liver and other steroidogenic
tissues such as the adrenal gland, ovaries, and testes
through the interaction of HDL with the SR-B1 recep-
tor. Alternatively, the cholesteryl esters in HDL can be
exchanged for the triacylglycerols in VLDL and IDL
through the action of cholesteryl ester transfer protein
(CETP). Such a cholesteryl ester is eventually re-
moved by the interaction of IDL or LDL with the
LDL receptor on the surface of liver or peripheral
cells.
Important Enzymes Responsible for the
Processing of Lipoprotein Particles in the
Bloodstream and Peripheral Tissues
0020Apolipoproteins present on the surface of lipid
particles serve as ligands for receptors as well as
activators or inhibitors of enzymes important in the
lipoprotein-processing pathway. The names and func-
tions of enzymes playing a major role in the process-
ing of lipid particles are summarized in Table 2.
Functions of some of the important apolipoproteins
and enzymes will be discussed briefly.
0021LPL catabolizes the triacylglyceride in the core of
many lipoprotein particles. LPL converts chylo-
microns into chylomicron remnants, and VLDL par-
ticles into VLDL remnants and IDL. LPL is bound to
heparan sulfate proteoglycans on the surface of capil-
laries, and production of this enzyme is upregulated
by insulin. Heparinase-induced proteoglycan defi-
ciency (heparinase cuts off proteoglycans from the
cells) impairs the function of LPL. Both chylomicrons
and chylomicron remnants are cleared from the
bloodstream by the liver through the binding of the
ligand, apoE, either to the LRP receptor or to the LDL
(B,E) receptor. This initiates internalization via
coated pits and lysosomal degradation of the apoE-
coated lipoprotein particles.
0022LCAT is made in the liver and is present on HDL
particles. Initially, HDL has very little cholesteryl
ester but is loaded up with cholesteryl ester by the
action of LCAT. This HDL particle then undergoes
exchange of cholesteryl esters for triacylglycerol on
the triacylglycerol-rich lipoproteins through the
action of the CETP. The triacylglycerol-rich lipopro-
teins are then processed by HL, present on the surface
of liver cells. HL hydrolyzes triacylglycerols and
phospholipids. HL is similar to LPL and belongs
to the lipase superfamily. Both enzymes, CETP and
HL, are needed for reverse cholesterol transport as
1232 CHOLESTEROL/Absorption, Function, and Metabolism
demonstrated by transgenic experiments/substitution
experiments. Estrogen is an important negative regu-
lator of HL. Phospholipid transfer proteins (PTP) are
made in multiple tissues. PTPs play an important role
in the maintenance of plasma HDL content and
remodeling of HDL in the circulation.
Role of Transcription Factors in the
Regulation of Cholesterol Metabolism
0023 Recently, transcription factors (proteins that bind to
DNA and control transcription of proteins important
in lipid metabolism) have been described that tightly
control the amount of cholesterol present inside the
cell.
0024 Uptake of cholesterol in liver cells is mediated by
receptors such as the LRP receptor and the LDL
receptor. Brown, Goldstein, and coworkers have de-
scribed transcription factors called sterol regulatory
element-binding proteins (SREBPs). SREBPs control
the transcription of enzymes such as HMG CoA
reductase, essential in cholesterol biosynthesis, and
of the LDL receptor, which mediates the uptake
of LDL by the liver. It has been known for years
that cholesterol in the diet shuts down the synthesis
of new cholesterol in the liver. It does so by down-
regulating the rate-limiting enzyme in the biosyn-
thesis of cholesterol, HMG CoA reductase. It is now
known that an increase in hepatic cholesterol levels
inhibits the sequential release of an active form of
SREBP by two proteases from the nuclear membrane.
Conversely, a decrease in hepatic cholesterol levels
promotes the proteolysis and release of SREBP,
allowing its translocation into the nucleus where
SREBP binds to the promoter of the LDL receptor
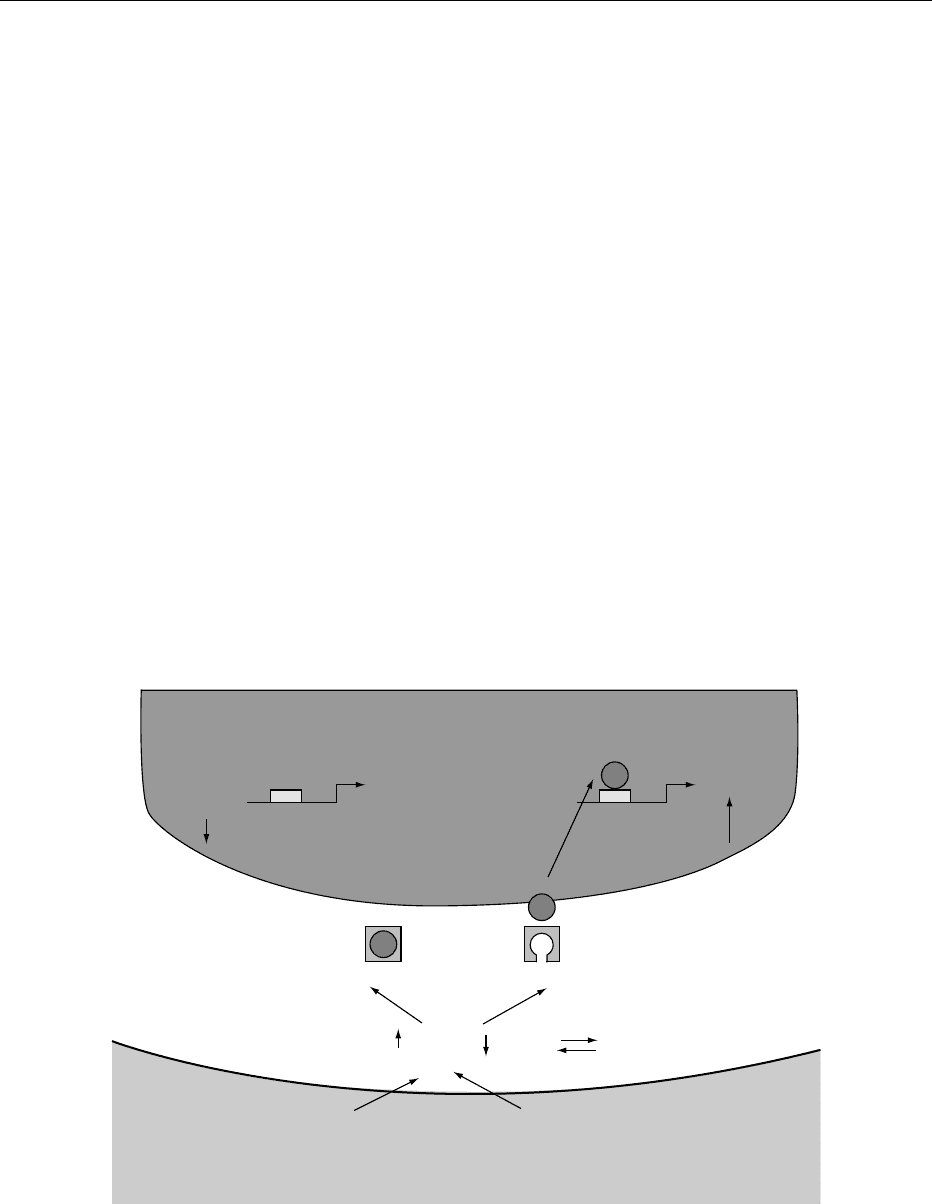
gene and activates its transcription (Figure 4). The
increased production of the LDL receptor molecule
results in an increased uptake of LDL cholesterol by
the cells. Also, SREBP interacts with the gene for
HMG CoA reductase, enhancing its transcription
and increasing the number of enzyme molecules.
This increases the biosynthesis of cholesterol. Thus,
both an increased uptake of cholesterol on LDL from
the blood and enhanced production of cholesterol
synthesis occur when the level of cholesterol in the
liver is decreased. Conversely, as the cholesterol pool
in the liver is increased, the release of SREBP is in-
hibited, the LDL receptor gene and HMG CoA reduc-
tase genes are downregulated, and an equilibrium is
achieved (see Figure 4).
X
LDL receptor LDL receptor
Nucleus
SREBP response element
Nuclear membrane
Decreased proteolysis of SREBP Increased proteolysis of SREBP
Cell membrane
Cholesterol
Cholesterol Cholesterol esters
Fatty acids
ACAT
fig0004 Figure 4 LDL-receptor/SREBP pathway. Liver cells adsorb cholesterol and fatty acids, and the amount of cholesterol versus
cholesterol ester in the cell cytoplasm is controlled by the enzyme acyl cholesterol acyl transferase (ACAT). If the level of free
cholesterol is decreased (on the right), a proteolytic process is activated to free the steroid response element-binding protein (SREBP)
which then translocates to the cell nucleus and activates the transcription of the LDL-receptor gene. This leads to an increased
expression of LDL-receptors on the surface of the liver cell and to enhanced clearance of LDL from the bloodstream. The reverse is
true (on the left) if the intracellular cholesterol level is increased.
CHOLESTEROL/Absorption, Function, and Metabolism 1233

0025 Other enzymes, such as the intracellular ACAT,
also play a role in this regulatory pathway; for
example, as ACAT converts free cholesterol into
cholesteryl esters, less unesterified cholesterol is avail-
able to inhibit the proteolysis of SREBP, leading to
increased release of SREBP and the upregulation of
both the LDL receptor and HMG CoA reductase. If
ACAT is less active, as might occur in the presence of
saturated fats (which are not preferred substrates for
the enzyme), more free cholesterol will be available to
inhibit the release of SREBP, leading to a downregu-
lation of LDL receptors and HMG CoA reductase. If
the inhibition of the LDL receptors is greater than the
inhibition of HMG CoA reductase, as occurs in about
one in four humans, an increase in the blood choles-
terol and LDL cholesterol occurs.
0026 Another important protein involved in the control
of cholesterol synthesis is the SREBP cleavage acti-
vation protein (SCAP). The NH
2
-terminal protein
domain of SCAP resembles the NH
2
-terminal pro-
tein domain of HMG CoA reductase, and it is
thought that both function as sterol-sensing domains.
SCAP is a required activator of SREBP cleavage, as
cleavage is usually abolished in the presence of
sterols. The family of SREBP transcription factors
also appears to be important in the regulation of
fatty acid synthesis. acetyl CoA carboxylase, fatty
acid synthase, stearyl CoA desaturase-1 (a necessary
enzyme for the generation of unsaturated fatty acids),
and even the enzyme LPL appear to be modulated by
SREBP.
Inherited Mechanisms of Disorders of
Lipid and Lipoprotein Disorders
0027 Apolipoproteins are very important in cholesterol
metabolism as they act as enzymes and ligands for
receptors mediating lipid modification and absorp-
tion (see also above). Most apolipoproteins are
made in the liver and intestine, but a small amount
of apoB can be made in heart tissue and apoE is made
by macrophages. All tissues can degrade apolipo-
proteins, but most of them are degraded in the liver.
0028 One of the most serious inherited abnormalities
of the cholesterol metabolism is a condition called
familial hypercholesterolemia (FH). In FH, the LDL
receptor is defective, and liver cells have a reduced
capacity to bind and take up LDL cholesterol from
blood. FH can occur in a severe homozygous form
where patients have few, if any, functional LDL recep-
tors. Homozygotes for FH are rare, about one in a
million, whereas patients with one mutant allele and
one normal allele for the LDL receptor, FH heterozy-
gotes, occur in about one in 500 persons. The ligand
for the LDL receptor, apoB-100, on the surface of
LDL particle can also be defective, and this may also
lead to inherited high total cholesterol and LDL
cholesterol levels similar to those with FH.
0029More than 150 mutations in the LDL receptor gene
have been described, and grouped into five different
classes: the class I mutations do not permit the LDL
receptor to be synthesized; class II mutations interfere
with the transport of the receptor to the surface of the
cell; class III mutations produce a receptor that does
not bind normally to its ligand, apoB-100; in class IV
mutations, the LDL receptor binds to LDL but it can
not be internalized; and in class V mutations, the
receptor cannot be reused by recycling.
0030Treatment of the homozygous FH individuals is
problematic, since it is difficult in many patients to
induce functional LDL receptors, either with a statin
or with a bile acid sequestrant. Niacin and a high dose
of statins may decrease the production of LDL by
inhibiting to a degree the formation and secretion of
VLDL in liver. FH homozygotes often die from
ASCVD before the age of 20 years. The atheroscler-
osis also affects the aortic valve and can lead to life-
threatening aortic stenosis. Transplantation of a liver
with functional LDL receptors was once used but has
fallen out of favor because of the significant morbid-
ity and mortality associated with this procedure. LDL
phoresis can often decrease the LDL levels by half;
however, the procedure must be repeated every 2
weeks, and sufficient blood flow can be a problem
in very young FH homozygoyes. There has been a
strong interest in gene therapy as a potential success-
ful treatment, but it is still experimental. Heterozy-
gous FH individuals can be successfully treated by a
combination of diet, exercise, and drugs.
Treatment of Lipid Disorders
0031Lipid disorders have traditionally been classified
according to the increased amount of lipoprotein
particles present in blood (Fredrickson’s class I–V,
Table 3).
0032With increasing knowledge, mutations in recep-
tors, apolipoproteins, and enzymes have been identi-
fied as the cause of these abnormalities, but many
defects are still not known, and often, their expres-
sion is modulated by environmental and genetic
factors as well. Many lipid disorders are secondary
to other diseases. For example, the LPL is upregulated
by insulin, and diabetic patients who do not produce
enough insulin often manifest a hypertriacylglycerol-
emia secondary to their diabetes and relative LPL
deficiency. Other medical conditions such as hypo-
thyroidism can cause an elevated LDL cholesterol
level, and in such cases, the other medical condition
must be treated first. Certain classes of drugs can also
1234 CHOLESTEROL/Absorption, Function, and Metabolism

contribute to lipid abnormalities and, if so, can be
discontinued if medically feasible.
0033 Elevated LDL-cholesterol levels are strongly correl-
ated with development of ASCVD, whereas a high
HDL-cholesterol level is usually protective against
ASCVD. The National Cholesterol Educational Pro-
gram (NCEP) uses measurements of LDL-cholesterol
as guidelines for treatment with diet and drug
therapy in addition to multiple risk factors (Tables 4
and 5).
0034The first step in treatment of a primary disorder of
lipid and lipoprotein metabolism is a diet reduced in
total fat, saturated fat, and cholesterol (NCEP step I
and step II diets; see Table 6). Weight loss is particu-
larly important, especially in those with hyper-
triacylglycerolemia, low HDL cholesterol, glucose
intolerance, and insulin resistance. Drug treatment is
usually based on the particular lipoprotein that is
elevated starting with the atherogenic LDL. For
every individual, positive and negative risk factors
as outlined by the NCEP III guidelines determine
whether the aim of treatment with diet, exercise, and
drug treatment has been reached (Tables 4 and 5).
Guidelines have also been proposed for hypertriacyl-
glycerolemia and low HDL cholesterol. A plasma
triacylglycerol level in adults above 400 mg dl
1
is
elevated, 200–400 mg dl
1
is borderline, and below
200 mg dl
1
is normal. An HDL cholesterol less than
40 mg dl
1
is too low.
0035The drugs of choice to treat an elevated LDL chol-
esterol level are the HMG CoA reductase inhibitors,
or the so-called ‘statins.’ Inhibition of this enzyme
causes a reduction in the hepatic cholesterol pool,
leading to an induction of LDL receptors and a fall
in blood LDL cholesterol levels (see above). In 1976,
a fungal metabolite, called mevinolin or mevastatin,
was isolated, which inhibited HMG CoA reductase.
Most of the currently available cholesterol-lowering
drugs, which target these enzymes, are chemically
modified fungal metabolites or completely synthetic
analogs of this compound. These include: atorvasta-
tin, fluvastatin, lovastatin, pravastatin, and simvasta-
tin. When given in sufficiently high doses, these drugs
can also produce a significant fall in triacylglycerols,
presumably by decreasing hepatic VLDL production
and by removing the IDL molecules. HDL levels often
increase with statin therapy, but the mechanism is not
currently known.
0036Other drugs that are given when the LDL choles-
terol level is elevated are the bile acid sequestrants or
resins (cholestyramine or colestipol) and niacin.
tbl0003 Table 3 Classification of lipid disorders by phenotype (after Fredrickson)
Group Lipoproteinin excess Plasma cholesterol Plasma triacylglycerol Plasma appearance
I Chylomicrons Elevated or very elevated Very elevated Clear plasma with creamy layer on top
IIa LDL Elevated Normal Clear
IIb LDLþVLDL Elevated Moderately elevated Clear, turbid
III b-VLDL, IDL Elevated Elevated Turbid
IV VLDL Normal or moderately elevated Elevated Turbid
V ChylomicronsþVLDL Elevated or very elevated Very elevated Turbid plasma, creamy layer on top
tbl0004 Table 4 Intervention National Cholesterol Education Program
(NCEP) guidelines
LDL-cholesterollevelwith Initiation of diet Addeddrug therapy
Less than two risk factors >160 mg dl
1
>190 mg dl
1
More than two risk factors >130 mg dl
1
>160 mg dl
1
ASCD >100 mg dl
1
Diabetes mellitus >100 mg dl
1
Patients with diabetes mellitus or ASVD have a treatment goal for LDL
cholesterol of equal or less than 100 mg/dl independent of the presence or
absence of risk factors.
From National Cholesterol Education Panel (NCEP) (1993) Adult treatment
panel II. Journal of the American Medical Association 269: 3015–3023, with
permission.
tbl0005 Table 5 Atheroclerotic vascular disease (ASVD) risk factors
after the National Cholesterol Education Program (NCEP)
guidelines
Positive
risk factor
Negative
risk factor
HDL cholesterol value <40 mg dl
1
>60 mg dl
1
Age Men >45 years of age
or older
Women >55 years of age
or older
Family history of
premature ASVD
Present
Current cigarette
smoking
Present
Hypertension Present
Each person’s risk factors are to be determined and used as a guideline
for treatment. Patients with diabetes mellitus or ASVD have a treatment
goal for LDL cholesterol of equal or less than 100 mg dl
1
independent of
the presence or absence of risk factors.
From National Cholesterol Education Panel (NCEP) (1993) Adult treatment
panel II. Journal of the American Medical Association 269: 3015–3023, with
permission.
CHOLESTEROL/Absorption, Function, and Metabolism 1235