Bongard Frederic , Darryl Sue. Diagnosis and Treatment Critical Care
Подождите немного. Документ загружается.


RESPIRATORY FAILURE
297
and lymphocytes and their cytokines, prostaglandins,
leukotrienes, platelets, coagulation factors, adhesion mole-
cules, and immunoglobulins, as well as exogenous substances
such as endotoxin and other products of bacteria and
fungi. Endogenous cell products have received the most
attention—some as mediators of injury, such as oxygen rad-
icals and proteolytic and elastolytic enzymes, but others as
amplifiers of inflammation and injury, such as interleukins,
platelet-activating factor, complement, and other substances
that are chemotactic, bronchoreactive, or vasoreactive. These
may be active in the early, middle, or late phases of lung
injury. A role for injury from oxygen radicals is supported by
the finding of reduced alveolar fluid glutathione in patients
with ARDS. The coagulation system has been suggested by
some investigators as having a central role in lung injury, per-
haps by linking intravascular events to direct injury to the
endothelium and by activation of inflammatory sequences.
Elevated plasma levels of tumor necrosis factor (TNF) are
found in some patients with ARDS but also in those with
sepsis and other systemic disorders. A potent cytokine, TNF
has a variety of systemic effects, some of which could cause
or potentiate lung damage. The finding of several elevated
cytokines in ARDS suggests the possibility of common regu-
latory factors being involved. One factor, NF-κB, regulates
production of TNF, interleukin 1 (IL-1), IL-6, and IL-8. This
hypothesis is attractive because of the frequent association of
IL-6, TNF, and IL-8 with lung injury. More recently, ARDS
has been linked to toll-like receptors (TLRs), which respond
to a variety of substances to trigger a vast cytokine response.
TLRs are responsible for innate immunity; this could
explain the common finding of ALI in response to the range
of inciting factors.
A key role for polymorphonuclear leukocytes in lung
injury is supported by finding neutrophils in large numbers
in the lungs of ARDS patients. Furthermore, neutrophils are
primed to release potentially toxic substances from their
granules, neutrophil chemotactic factors and activators are
increased, and in some animal models, lung injury is attenu-
ated after neutrophil depletion. For example, neutrophil-
activating protein/interleukin-8 (NAP-1/IL-8) has been
found in high concentrations in alveolar fluid, and there was
a correlation with the number of neutrophils. High concen-
trations of NAP-1/IL-8 also were associated with poor clinical
outcome. On the other hand, neutropenic cancer patients
may develop ARDS indistinguishable from that observed in
nonneutropenic patients, and diffuse alveolar damage in
some animal models does not require the presence of neu-
trophils. Levels of both cytokines and modulators of cytokine
function are highly variable in ARDS, and it is clear that
cytokines taken individually or as patterns of response are not
able to predict development or prognosis of ARDS. In paral-
lel with the diversity of clinical conditions associated with dif-
fuse alveolar damage and ALI, it is highly likely that different
conditions in different patients explain why consistent find-
ings cannot be identified. While this makes a single common
causative mechanism unlikely, this hypothesis helps to
explain why so many conditions can result in very similar his-
tologic and physiologic features. Nevertheless, there is now
ample evidence that persistent elevation of inflammatory
cytokines in blood or alveolar fluid is associated with poor
outcome in ARDS in all forms of this disorder.
Patients may develop secondary bacterial or fungal pneu-
monia during the course of ARDS, further confusing the pic-
ture. Administration of high concentrations of inspired oxygen
contributes to lung injury, and high airway pressure and rela-
tively high tidal volume during mechanical ventilation are
closely linked to worsening pulmonary edema and fibrosis. On
the other hand, higher oxygen requirements and airway pres-
sures may simply indicate more severe underlying disease.
B. Noncardiogenic Pulmonary Edema—Normal lungs are
kept very dry to permit efficient gas exchange, and the struc-
ture and activity of the lungs maintain only a small amount
of fluid in the lungs. Normal lungs have tight junctions
between alveolar epithelial cells, an extensive lymphatic sys-
tem, low hydrostatic pressure in the pulmonary capillaries,
and other mechanisms to avoid pulmonary edema. Thus,
lung injury from any number of insults can promote pul-
monary edema by damaging these mechanisms.
Pulmonary edema is a major clinical manifestation of
ARDS, and the pulmonary edema fluid contains a high con-
centration of protein. This is in marked contrast to pul-
monary edema owing to elevated pulmonary venous
pressure (hydrostatic pulmonary edema) or to decreased
plasma albumin concentration (hypo-oncotic pulmonary
edema), in which the edema fluid is a low-protein transu-
date. ARDS also has been called exudative or noncardiogenic
pulmonary edema, reflecting the increased permeability of
the injured lung to water, solute, and protein. Exudative pul-
monary edema forms in the absence of elevated pulmonary
artery wedge pressure, and the ratio of edema fluid protein to
plasma protein is high. Edema fluid accumulates both in the
pulmonary interstitium and in the alveoli, and because of
potential fluid pathways, lung lymphatics and bronchovascu-
lar spaces (surrounding the bronchioles, bronchi, and pul-
monary arteries) may become engorged. Pulmonary edema
removal by the pulmonary circulation and lymphatics,
including active transport of solute and water, is severely
impaired because of the ALI. In a minority of ARDS patients,
the pulmonary epithelium is able to resolve pulmonary
edema during the first 12 hours. This probably reflects rela-
tively preserved epithelial cells that might increase solute and
water transport in response to β-adrenergic agonists. These
and other drugs are currently being studied.
C. Chronic Lung Injury—Diffuse alveolar damage seen in
ARDS may follow several courses, including resolving
entirely with little or no evidence of chronic damage after
weeks or months. However, other patients develop mild to
severe pulmonary fibrosis. One of the most interesting find-
ings in ARDS is evidence of very early deposition of type III
collagen (procollagen III peptide in alveolar fluid) in the
lung, sometimes within 24 hours of the onset of diffuse
CHAPTER 12
298
alveolar damage. Evidence for early fibrosis has been associ-
ated with poor prognosis, stressing the potentially inappro-
priate role of remodeling and repair in late lung injury. These
findings have challenged the time course of ALI, with irre-
versible fibrosis occurring much sooner than in earlier pro-
posed models. Attraction and activation of fibroblasts may
be mediated by various substances such as platelet-activating
factor that are increased in blood and pulmonary edema
fluid. Oxygen toxicity may play a role, especially if patients
require very high inspired oxygen to treat hypoxemia.
Another potential contributor to chronic lung damage is rec-
ognized to be overdistention of the lungs by high tidal vol-
ume, high PEEP, or high airway pressure.
In chronic lung injury, variable amounts of collagen are
laid down into the alveolar and interstitial spaces with distor-
tion and disruption of the normal lung parenchyma, result-
ing in restrictive lung disease, reduced exercise capacity, and
hypoxemia. Histologic findings may be indistinguishable
from idiopathic pulmonary fibrosis. Particularly sensitive
findings are the Pa
O
2
and P(
A
–a)
O
2
during exercise. Recent
data show that survivors with more severe early ARDS had
worse late-stage pulmonary function than those with less
severe acute disease. Lung function as assessed by spirometry,
total lung capacity, and diffusing capacity for carbon monox-
ide averaged about 80% of predicted in these survivors.
The greatest degree of improvement was seen in the first
3 months, and there was little additional improvement
between 6 months and 1 year.
The relationship of collagen deposition, severity of lung
injury, and outcome provides some possibilities for interven-
tion. These include potential inhibition of chemotactic or
activating factors for fibroblasts, removal of some forms of
collagen during tissue repair phases, and limitation of lung
injury by controlling inflammation, oxygen toxicity, and
ventilator-induced lung injury.
D. Physiologic Manifestations
1. Refractory hypoxemia—Hypoxemia in ARDS is due to
right-to-left shunt and
.
V/
.
Q mismatching resulting from
atelectasis and filling of alveolar spaces with edema fluid.
.
V/
.
Q mismatching also may result from nonuniform
changes in airway resistance, decreases in regional lung
compliance, and primary and secondary alterations of
lung blood flow. Hypoxemia usually is severe and not cor-
rected even when the patient is given high concentrations
of inspired O
2
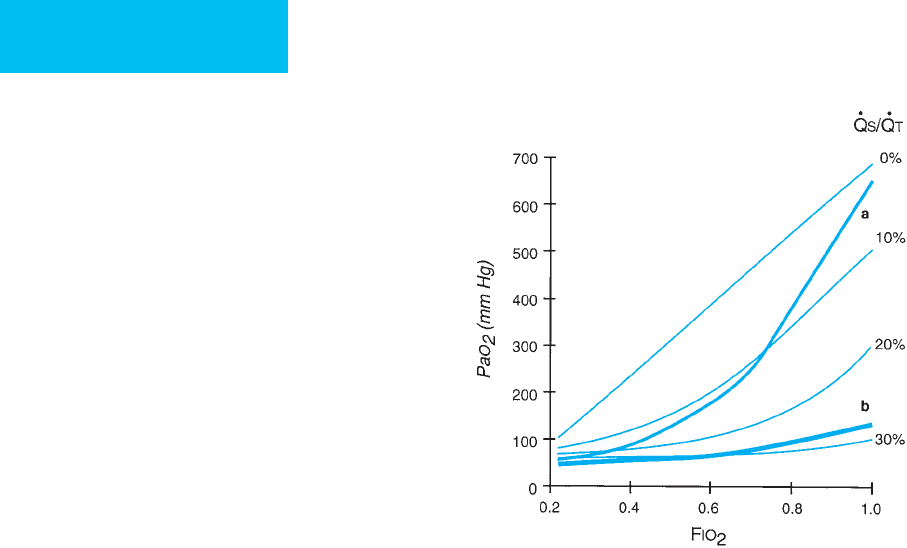
,termed refractory hypoxemia (Figure 12–7).
As part of the definition of ARDS, the ratio of Pa
O
2
to F
IO
2
is less than 200.
As shown by CT scanning, the majority of lung in ARDS
is completely airless, with small proportions either normally
inflated or collapsed with potential for recruitment. These
findings suggest that right-to-left shunting plays the major
role in refractory hypoxemia, whereas the use of PEEP walks
a fine line between recruitment of atelectatic lung and
overdistention of normal lung.
2. Altered static lung mechanics—Lung compliance is
severely decreased and airway resistance mildly increased in
ARDS. Decreased lung compliance results from a combina-
tion of interstitial pulmonary edema, collapse of lung units,
airway obstruction, and inactivation of alveolar surfactant.
Ineffective surfactant in ARDS may be due to reduced pool
sizes, alteration in surfactant proteins, altered metabolism,
or inactivation by plasma proteins, oxygen radicals, or
phospholipases exuding into the alveolar spaces. In later
stages, lung compliance is reduced because of accumulation
of collagen.
Studies correlating regional radiographic lung volume
change and inflation pressure show that disease involvement
in ARDS is much less uniform than formerly thought. This
finding has resulted in a major change in understanding of
ARDS. Most lung regions are extensively involved and com-
pletely airless, some participate variably in gas exchange, and
other uninvolved regions accept the bulk of ventilation.
These latter regions have normal specific lung compliance,
indicating that the primary cause of decreased overall lung
Figure 12–7. Lines showing Pa
O
2
versus F
IO
2
for vari-
ous constant-shunt fractions (
.
Q
S
/
.
Q
T
) between 0% and
30%. Superimposed are typical responses in patients with
ARDS (lines a and b). Line b shows severe hypoxemia
with a response suggesting severe right-to-left shunt as
the primary mechanism of hypoxemia. Line a demon-
strates severe hypoxemia but with Pa
O
2
increasing more
rapidly with increasing F
IO
2
than with pure right-to-left
shunt. The mechanism of hypoxemia is probably severe
ventilation-perfusion mismatching.
RESPIRATORY FAILURE
299
compliance is overdistention of these small uninvolved
regions of the lung rather than uniform stiffening of the
entire lung. This finding has major implications for how
patients with ARDS should be mechanically ventilated,
notably the use of a low-tidal-volume strategy.
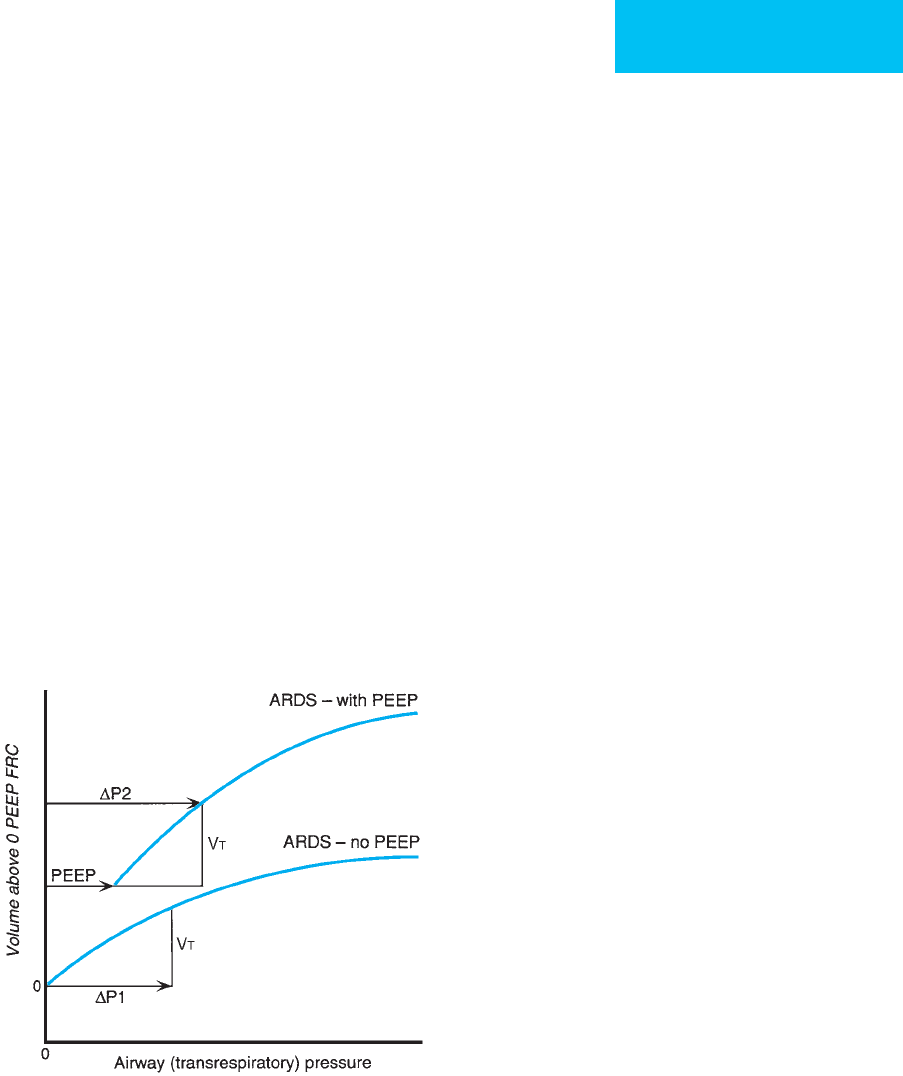
The pressure-volume (PV) curve of the lungs in ARDS is
shifted downward and rightward (Figure 12–8). The lungs
require greater pressure to inflate, and the work of breathing
is increased. An increase in lung compliance may indicate
improvement of the disease or recruitment of atelectatic
lung, especially with the application of PEEP. Some investiga-
tors have noticed that the respiratory system compliance
curve, which considers both lung and chest wall mechanics,
is altered in ARDS. Patients with nonpulmonary causes of
ARDS (eg, abdominal sepsis, trauma, or postoperative) had
greater response to PEEP, suggesting that the “pulmonary
ARDS” patients (mostly those with pneumonia) had more
severe and less recruitable lung consolidation, whereas the
nonpulmonary ARDS patients had more atelectasis.
The shape of the PV curve has been stressed by some cli-
nicians. The curve has been divided into sections, including
a flat initial increase in volume with increasing pressure, a
lower inflection point after which compliance (slope)
increases, an upper inflection point, and then another region
of low compliance (flat slope). The regions of the PV curve
may have implications for adjusting PEEP (see below) and
limiting tidal volume.
3. Increased airway resistance—Increased airway resist-
ance is described in ARDS patients, probably owing to edema
in the bronchovascular spaces surrounding the bronchi, but
there may be inflammatory mediators that induce bron-
choconstriction. Another cause may be the normal increase
in airway resistance in areas of decreased pulmonary perfu-
sion in response to ventilation-perfusion mismatching. The
increased airway resistance, as much as sixfold compared
with normal individuals, contributes to higher airway pres-
sure and work of breathing. In one study, increased resistance
correlated positively with both peak airway pressure and the
severity of gas-exchange abnormality.
E. Multiple-Organ-System Failure—Although often viewed
as a primary lung disorder, ARDS is clearly associated with
multiple-organ-system dysfunction and failure. Subtle evi-
dence of organ-system dysfunction is very common in both
survivors and nonsurvivors of ARDS, and renal failure, liver
failure, CNS failure, heart failure, thrombocytopenia, and GI
bleeding and malabsorption contribute to mortality and mor-
bidity. In fact, of those who die with ARDS, respiratory failure
has been estimated to be the primary cause in as few as 16%.
Sepsis and nonrespiratory failure accounted for the remainder.
The cause of multiple-organ-system failure in ARDS
remains poorly understood. There are three major theories.
First, some investigators believe that the same systemic
process that damages the lungs injures other organs as well;
ARDS is simply the most obvious and earliest manifestation.
For example, one study found that ARDS patients had
increased urinary myoglobin and β
2
-microglobulin during
development of pulmonary edema, and there was a correla-
tion between pulmonary edema and the amount of these
proteins in the urine. This mechanism of multiple-organ-
system failure may be particularly likely in patients with sep-
sis and shock. Circulating factors such as endogenous
cytokines or endotoxin are likely mediators of multiple-
organ-system failure in this hypothesis.
Another hypothesis is that hypoxemia and inadequate O
2
delivery are the causes of multiple-organ-system failure.
Some studies of ARDS patients suggest that tissue oxygen
consumption depends on systemic oxygen delivery even
when oxygen delivery is normal or elevated. Thus a small
decrease in oxygen delivery can cause oxygen consumption
to fall, resulting in tissue hypoxia and potential organ dam-
age. Studies undertaken to test the hypothesis that increasing
oxygen delivery will raise oxygen consumption, however,
have not shown improved outcomes.
A final consideration in multiple-organ-system failure is
the effects of therapy of ARDS. Positive-pressure ventilation
and PEEP are important parts of the supportive care of
ARDS, but these can have adverse effects on cardiac output
and oxygen delivery. Invasive monitoring, artificial airways,
and other devices increase risk of infection and sepsis. This
theory is attractive because mortality in ARDS, while still
Figure 12–8. Hypothetical respiratory system pressure-
volume curves for a patient with ARDS showing a flatter
than normal relationship (decreased respiratory system
compliance, C
rs
= V
T
/ΔP
1
). With addition of PEEP, a shift to
a more compliant curve may occur such that C
rs
= V
T
/
(ΔP
2
− PEEP) increases. The change in compliance may
represent recruitment of poorly ventilated or nonventi-
lated lung units with application of PEEP and may be
correlated with improved oxygenation and gas exchange.

CHAPTER 12
300
high, is less often related to respiratory failure than to non-
respiratory organ dysfunction and sepsis. Recent clinical
studies provide circumstantial support for this theory. For
example, use of a low-tidal-volume strategy reduces mortal-
ity in ARDS patients by close to 25%. Because there was no
difference in the degree of obvious barotrauma, a beneficial
effect on nonrespiratory organ system function might be one
explanation.
Clinical Features
A. Predisposing Conditions—More than 100 clinical situa-
tions have been associated with development of ARDS.
Prospective studies have helped to identify the most com-
mon conditions, and 60–80% of cases of ARDS can be
accounted for by sepsis, trauma, diffuse pulmonary infec-
tion, and aspiration of gastric contents, with other condi-
tions being much less frequent (Table 12–18).
The overall attack rate of ARDS is relatively low. In one
study, only 8% of patients at risk with any of the nine most
frequently associated predisposing conditions developed
ARDS, although the incidences ranged from 6–50% for indi-
vidual risks. In those with multiple predisposing conditions,
the incidence of ARDS increases substantially, with an aver-
age incidence of 42% for two or more risks for ARDS com-
pared with 19% for those with a single risk in one
study—although 41% of patients with sepsis alone devel-
oped ARDS.
1. Sepsis—Sepsis is present in as many as 50% of ARDS
patients. In medical patients, sepsis is the most common
ARDS association; in trauma patients, sepsis is the most
common association in ARDS developing 48 hours or more
after admission. A distinction sometimes can be made
between pneumonia leading to ARDS and infection from a
nonlung site leading to ARDS. In severe pneumonia, gas-
exchange abnormalities and chest x-ray features of ARDS are
sometimes seen prior to or in the absence of other character-
istics of systemic infection, suggesting that much of the lung
injury is due to the infectious agent and local response to
infection within the lung. Pathogens include bacteria,
mycobacteria, fungi, viruses, Pneumocystis jerovici, and rick-
ettsiae. On the other hand, a nonpulmonary primary site of
infection is often identified in ARDS patients, including the
GI tract, urinary tract, heart, or soft tissues. Although gram-
negative enteric bacilli are encountered most often, ARDS
can result from systemic infection with any bacterial, viral, or
fungal pathogen. Sepsis is of particular interest because it
appears that circulating mediators, including interleukins
and other responses to infection, are largely responsible for
ARDS, hypotension, and multiple-organ-system failure, in
addition to the microbial organisms themselves. Sepsis com-
plicated by ARDS has a higher mortality, especially if the
source of infection cannot be identified and treated readily.
2. Aspiration of gastric contents—Aspiration of gastric
contents is defined as an observed aspiration during intuba-
tion or witnessed vomiting in a patient with impaired airway
protective mechanisms. Although acidic gastric fluid is
thought to be the primary cause of lung injury and ARDS,
studies support a contributory role for bacteria, partially
digested food particles, gastric enzymes, and other noxious
substances. Neutralization of gastric acid prior to aspiration
does not prevent or moderate ALI. Other syndromes of aspi-
ration, including necrotizing pleuropulmonary infection and
lung abscess, can lead to ARDS if there is severe lung injury
response or sepsis. Aspiration of gastric contents is particu-
larly common in patients of advanced age, those who have
neurologic diseases resulting in paralysis or impaired swal-
lowing, or those who have advanced organ-system failure. In
other ICU patients, including surgical and obstetric patients,
aspiration potential is increased by sedatives, muscle relax-
ants, general anesthesia, and local anesthesia to the pharynx
and larynx; during endotracheal intubation; during enteral
feeding; and in diabetics or others with impaired GI motility.
In a sizable number of patients, aspiration of gastric contents
can only be presumed because predisposing factors are
absent and there is no observed aspiration event.
3. Trauma—Trauma is a common predisposing condition to
ARDS, but the precise mechanism is uncertain. Direct
trauma to the thoracic wall may result in lung contusion,
with hemorrhage into the lung causing abnormal gas
exchange, atelectasis, and further lung injury.
Nonthoracic trauma is also associated with an increased
incidence of ARDS. In some of these patients, lung injury
Table 12–18. Some predisposing conditions associated
with ARDS.
Infection
Pneumonia: bacteria, fungi, viruses,
Pneumocystis jerovici
Nonpulmonary:
Sepsis from gram-negative bacilli, staphylococci,
other gram-positive cocci,
Candida
Aspiration of gastric contents
Trauma
Thoracic: lung contusion
Nonthoracic
Hemorrhagic shock
Head trauma
Burns
Blunt abdominal trauma and pancreatitis
Orthopedic: fat embolism syndrome, severe fracture
Other conditions
Drugs: opiate or salicylate overdose
Pancreatitis
Toxic: smoke or gas inhalation
Amniotic fluid embolism
Central nervous system pulmonary edema
Near-drowning
Multiple transfusions of blood and blood products
Collagen-vascular disease, including vasculitis and pulmonary
hemorrhage

RESPIRATORY FAILURE
301
results from fat embolism syndrome, a disorder seen in frac-
tures of long bones, or pancreatitis from blunt or sharp
abdominal trauma. In others, the risk of ARDS correlates
with hypotension and shock and with the amount of blood
and blood products transfused, suggesting that the nature and
extent of trauma may have something to do with the develop-
ment of ARDS. Hypotension and shock release inflammatory
cytokines, and tissue damage liberates a variety of products
that could result in lung injury. Although the number of
blood transfusions administered seems to correlate with the
development of ARDS, the requirement for transfusions is
usually closely tied to the severity of trauma. Any patient,
however, who receives blood products has a risk of lung
injury owing to transfusion-related acute lung injury
(TRALI). While other mechanisms are present, TRALI is pri-
marily thought to be mediated by antibodies in donor plasma
reacting with recipient leukocytes. Blood products from mul-
tiparous donors have an increased risk of causing TRALI.
4. Risk modifiers—Cigarette smoking is associated with
increased likelihood of permeability pulmonary edema and
alveolar hemorrhage. With similar acute risk for ARDS, a
higher proportion of chronic alcoholics will develop ARDS.
Although risk balancing is difficult, elderly patients seem to
be at somewhat higher risk of developing ARDS, and out-
come has been reported to be poorer. Some studies have
shown that ARDS mortality is higher in men.
B. Symptoms and Signs—Patients have severe dyspnea and
respiratory distress. Findings include features of hypoxemia,
such as cyanosis, tachycardia, and tachypnea, but rales and
wheezes are the only features on chest examination, and
these are often surprisingly mild. Although the lungs are
filled with fluid, sputum production is rare, except in those
with bacterial pneumonia. Evidence of the underlying prob-
lem leading to ARDS may be found, including fever,
hypotension, trauma, or findings of organ-system dysfunc-
tion. Features of congestive heart failure are notably absent.
Early in ARDS, symptoms and signs are limited to the lungs.
If multiple-organ-system failure develops, then features of
hepatic, renal, or CNS failure may become evident.
C. Laboratory Findings—A key feature of ARDS is refrac-
tory hypoxemia. Pa
O
2
is severely reduced even when the
patient is given supplemental oxygen. Even 100% O
2
may not
raise Pa
O
2
above 60–100 mm Hg. Arterial pH may be high,
normal, or low depending on the success of the patient in
maintaining Pa
CO
2
with severe lung disease and the presence
of hypotension and metabolic acidosis.
Other laboratory findings reflect the clinical condition
leading to ARDS and the multiple-organ-system dysfunction
seen as a consequence of this disorder. Renal and hepatic fail-
ure and electrolyte disturbances are frequent complications.
D. Imaging Studies—At disease onset, the chest x-ray shows
diffuse bilateral infiltrates consistent with pulmonary edema.
The infiltrates range from patchy reticular to dense consoli-
dation. Unless there is coexisting heart disease, cardiomegaly
is absent, and there is a lack of the central perihilar promi-
nence of edema seen in congestive heart failure. In fact, some
have pointed out that ARDS is associated with predomi-
nantly peripheral distribution of infiltrates. Nevertheless,
distinguishing cardiogenic from noncardiogenic pulmonary
edema is never perfect. In patients with severe pneumonia
leading to ARDS, there may be focal densities in addition to
diffuse pulmonary edema. In later stages of ARDS, the origi-
nal dense opacification may change to a pattern of reticular
densities consistent with the proliferative and fibrotic stages
of lung injury. While chest x-rays usually suggest diffuse
involvement, marked nonhomogeneity of lung involvement
is seen on chest CT scans. Chest x-rays are essential in mon-
itoring patients for complications of ARDS, including baro-
trauma. Early evidence of air leaking into the lung
interstitium sometimes may be found as linear low-density
streaks of air surrounding bronchovascular bundles.
Occasionally, this finding is seen as a rounded lucent area
surrounding a pulmonary artery and bronchus. Air subse-
quently may track inward to the mediastinum (pneumome-
diastinum) or into the pleural space (pneumothorax).
Differential Diagnosis
Cardiogenic pulmonary edema is the most important dis-
order to be distinguished from ARDS (Table 12–19)
because treatment is often different. This may be particu-
larly difficult when ARDS is seen in conjunction with fluid
overload or concomitantly with congestive heart failure.
Septic shock may confound this distinction because circu-
lating endotoxin or cytokines may exert myocardial
depressant activity.
Table 12–19. Distinguishing cardiogenic from noncardio-
genic pulmonary edema.
Cardiogenic Noncardiogenic (ARDS)
Prior history of heart disease Absence of heart disease
Third heart sound No third heart sound
Cardiomegaly Normal-sized heart
Central distribution of infiltrates Peripheral distribution of infiltrates
Widening of vascular pedicle
(increased width of mediastinum
at level of azygos vein)
Normal width of vascular pedicle
Elevated pulmonary artery wedge
pressure
Normal or low pulmonary artery
wedge pressure
Positive fluid balance Negative fluid balance

CHAPTER 12
302
Treatment
Treatment of ARDS centers on management of severe hypox-
emia, correction of the underlying disease that led to ARDS,
and supportive care to prevent complications. Four major
treatment principles have evolved. First, almost all types of
therapy shown to benefit patients with ARDS—including oxy-
gen, PEEP, and positive-pressure ventilation—have potentially
severe adverse effects. Second, although ARDS is often consid-
ered primarily respiratory failure, multiple nonpulmonary
organ-system failure and infection are the major causes of
death in ARDS. Third, careful management of mechanical
ventilation, especially tidal volume, is associated with fewer
complications and is the only treatment demonstrated to
improve survival. Finally, prognosis is especially poor if the
underlying process is not identified or is poorly treated.
A. Oxygen—Treatment of hypoxemia in ARDS is begun
almost always using 100% oxygen (F
IO
2
= 1.0), and the con-
centration of O
2
is reduced with the goal of maintaining a
Pa
O
2
greater than 60 mm Hg (arterial O
2
saturation about
90%). Pa
O
2
increases only slightly with administration of
increasing concentrations of inspired oxygen (refractory
hypoxemia), even when 100% O
2
is given, indicating right-
to-left shunt or severe
.
V/
.
Q mismatching. A very few patients
can be managed using a nonrebreathing oxygen mask, but
most patients will be given oxygen via mechanical ventilation
because they are unable to tolerate the increased work of
breathing without mechanical support. The typical response
to administration of O
2
in ARDS is shown in Figure 12–7,
and examining the changes in venous admixture as O
2
and
other therapy are given can be helpful. The F
IO
2
should be
lowered as soon as possible to less than 0.5 to reduce the risk
of lung damage from oxygen toxicity. In most patients, low-
ering F
IO
2
is helped by using PEEP or other mechanical ven-
tilation methods to improve lung gas exchange. Because both
PEEP and high F
IO
2
both have the potential for complica-
tions, a compromise between high F
IO
2
and high PEEP often
must be chosen.
B. Positive End-Expiratory Pressure—PEEP includes both
positive end-expiratory pressure provided with mechanical
ventilation and continuous positive airway pressure (CPAP)
given to spontaneously breathing patients. During exhala-
tion without PEEP, alveolar pressure is higher than atmos-
pheric pressure, providing a pressure gradient, until
equilibrium is reached. When PEEP is applied, a pneumatic
valve terminates exhalation when the pressure in the system
decreases to a value set by the clinician. This PEEP is termed
extrinsic PEEP. The effective PEEP is the sum of extrinsic and
intrinsic PEEP (PEEPi).
1. Mechanism—The mechanism of action of PEEP is not
known, but PEEP probably works by counteracting the ten-
dency of alveoli to collapse in the face of pulmonary edema,
low lung volume, and loss of surfactant. Current understand-
ing of ARDS suggests that a majority of lung (although highly
variable) is completely atelectatic, whereas smaller propor-
tions are normally aerated or partially collapsed. PEEP
“recruits” some proportion of partially collapsed areas,
improving of gas exchange to these areas of low
.
V/
.
Q matching.
PEEP does not decrease the rate of pulmonary edema forma-
tion nor speed the rate of water reabsorption. It is also unlikely
that PEEP is able to open completely collapsed alveoli. Some
investigators have found that PEEP may affect the distribution
of pulmonary artery blood flow away from poorly ventilated
areas and toward better-ventilated regions, resulting in
improved arterial oxygenation. Finally, some studies have indi-
cated that PEEP may have a beneficial effect on the amount or
nature of lung injury.
If partially collapsed alveoli are recruited by PEEP to par-
ticipate in gas exchange, each tidal volume will be delivered
into a larger number of lung units. Lung compliance should
increase as PEEP as added, and the increase in compliance
should parallel improvement in arterial oxygenation (see
Figure 12–8). On the other hand, if PEEP simply distended
alveoli that are already participating in tidal gas exchange, then
lung compliance would remain constant or, if the lung units
become overdistended, lung compliance would decrease. An
upward shift in the position of the pressure-volume curve
indicates recruitment of lung units; movement along the orig-
inal pressure-volume curve suggests that no recruitment
occurred and that gas exchange will not be improved.
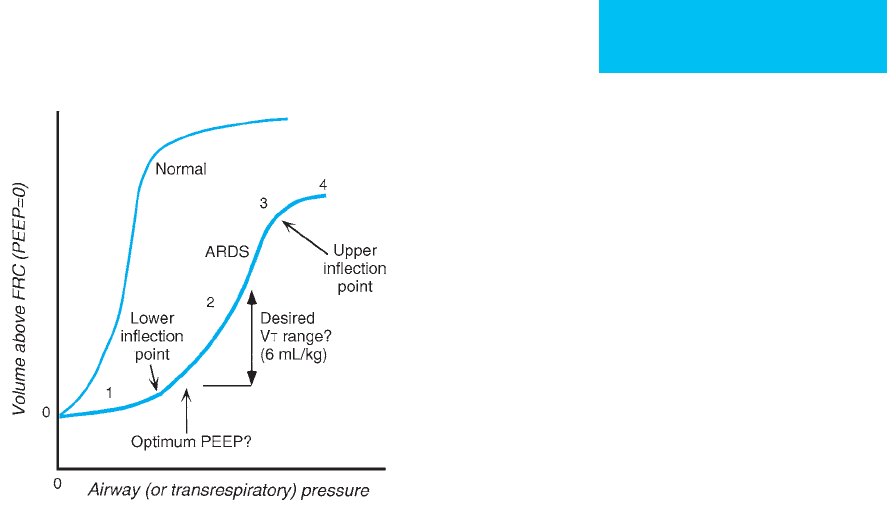
2. PEEP and the PV curve—In patients with early acute-
phase ARDS, the pressure-volume curve (Figure 12–9) is flat
at lung volumes near the end-expiratory volume. In this
region, compliance is abnormally low. As airway pressure is
raised—for example, during a tidal volume breath given with
positive-pressure ventilation—many patients will demon-
strate a region of steeper slope or higher compliance. In the-
ory, this region of higher compliance can only be explained by
recruitment of additional lung units. These units must have
been completely collapsed previously, but open when the air-
way pressure exceeds the units’ critical opening pressures. The
changeover point between low-compliance and higher-
compliance regions on the PV curve has been called the lower
inflection point. In theory, PEEP should be set just above the
lower inflection point to indicate recruitment.
Some investigators also have recommended giving suffi-
cient PEEP to prevent the patient breathing in the region cross-
ing the lower inflection point. At lung volumes near the
end-expiratory volume, alveoli are highly prone to collapse. As
the tidal volume is administered, alveoli are repeatedly exposed
to large shear stress as they change from completely closed to
completely open. This may lead to damage in the small airways
and bronchioles leading to the atelectatic lung units. Lung
injury may be moderated if sufficient PEEP is given to raise the
end-expiratory lung volume high enough to prevent cyclic lung
unit collapse (above the lower inflection point).
On the other hand, selecting an optimal PEEP value using
the lower inflection point remains controversial for several
reasons. First, some patients with ARDS have PV curves that
RESPIRATORY FAILURE
303
have no inflection point and no region of increasing compli-
ance. This has been associated with later stages of ARDS dur-
ing which lung units are poorly or not recruited even at
higher lung volume, and improvement of Pa
O
2
in response
to PEEP is generally poor. Second, some ARDS patients
whose respiratory PV curves have been partitioned into
lung and chest wall PV curves show no inflection point on
the lung PV curve but only on the chest wall PV curve. This
finding challenges the singular notion of lung recruitment
with PEEP. Next, the lower inflection point in ARDS patients
may be as high as 15–18 cm H
2
O, or considerably higher
than many clinicians believe is necessary. It is likely that a
more modest level of PEEP in conjunction with a low-tidal-
volume strategy will have the most physiologic and clinical
benefit. Finally, some workers have not found that the linear
segment of the PV curve is associated with constant increase in
compliance, implying that the lower inflection point is not
sharply defined.
3. Adverse effects—Adverse effects of PEEP include
reduced cardiac output that, in turn, decreases systemic oxy-
gen delivery. There have been several postulated mechanisms
of decreased cardiac output from PEEP, including, among
others, (1) decreased systemic venous return, (2) impaired
ventricular performance, (3) increased pulmonary vascular
resistance, and (4) decreased left ventricular compliance. It
is important to distinguish between decreased cardiac out-
put and decreased cardiac function, however, because
positive-pressure ventilation and PEEP can support ventric-
ular function, especially in severe pump failure and cardio-
genic shock. Decreased cardiac output lowers systemic
oxygen delivery, the product of cardiac output and arterial
oxygen content.
PEEP has been associated with barotrauma, including
pneumothorax and lung injury, and with exacerbation of
pulmonary edema, interstitial fibrosis, and inflammation.
Debate continues about whether the underlying lung disease
that led to the use of PEEP contributes to barotrauma or
whether barotrauma is solely related to PEEP. It is clear that
the degree of lung distention rather than the level of PEEP or
airway pressure is the important variable. In animal studies,
high end-inspiratory lung volumes were reached using either
low PEEP and high tidal volume or high PEEP and low tidal
volume. Both groups showed evidence of barotrauma. The
relationship of PEEP to barotrauma is clearly linked to tidal
volume, and the low-tidal-volume strategy will influence the
incidence of lung injury from PEEP.
Patients in whom PEEP is poorly tolerated have hypoten-
sion, tachycardia, and decreased cardiac output as the earli-
est manifestations. Hemodynamic intolerance may be signs
of volume depletion, severe pulmonary hypertension, or
ventricular dysfunction. A pulmonary artery catheter may be
helpful, and volume expansion or vasopressor drugs may be
necessary. In other patients, PEEP may improve Pa
O
2
only
slightly, suggesting that they have few recruitable lung units.
PEEP is generally contraindicated in very asymmetric or
localized lung disease with hypoxemia.
PEEP may paradoxically worsen Pa
O
2
in ARDS patients
with a patent foramen ovale. In 39 patients with ARDS given
PEEP of 10 cm H
2
O, mean P(
A
–a)
O
2
improved, and only 7
patients had an increase in shunt fraction, but in 7 patients
with patent foramen ovale, 6 had an increase in shunt frac-
tion and little or no reduction in P(
A
–a)
O
2
. Failure to
improve oxygenation with PEEP was likely due to increased
right-to-left shunting of blood through the foramen ovale,
owing to an increase in pulmonary vascular resistance medi-
ated by PEEP.
4. Application of PEEP—In patients with ARDS, many cli-
nicians prefer to give oxygen at F
IO
2
1.0 initially and then to
decrease F
IO
2
as long as adequate Pa
O
2
and O
2
saturation are
maintained. To do this, PEEP is titrated upward starting at 5
cm H
2
O. One strategy is to use the lowest predetermined
combination of PEEP and F
IO
2
(Table 12–20), with a clear
target for Pa
O
2
and O
2
saturation. Higher PEEP levels have
Figure 12–9. Hypothetical PV curves shown for normal
individuals and ARDS patients. Regions on the ARDS PV
curve include (1) a region of low compliance at low lung
volume—with a lower inflection point; (2) a region with a
steeper slope showing higher compliance—with an upper
inflection point; and (3) a region with a flatter slope
(poorly compliant). In theory, PEEP might be chosen to be
above the lower inflection point to maximize recruitment
and minimize shear stress on the lungs. Tidal volume
should be adjusted to stay within region (2) to avoid
overdistention. This concept has been challenged because
of evidence that recruitment occurs throughout the PV
curve and is not restricted to the area around the lower
inflection point. See text.
CHAPTER 12
304
not shown benefit, although studies of high versus low PEEP
have shown variable results. As described earlier, to avoid the
lowest lung volume range at which additional stress-induced
lung injury may occur, a minimum PEEP of 5 cm H
2
O prob-
ably should be given, but the minimum PEEP defined by the
lower inflection point may be substantially higher in some
patients. Recently, it has been suggested that the level of
PEEP could best be set by measuring the percentage of
recruitable lung. This avoids excessive PEEP in “nonre-
cruitable” patients but requires CT scanning of the lungs
while inflated to different pressures.
PEEP should be adjusted incrementally, with close moni-
toring of respiratory mechanics (especially inspiratory
plateau pressure), arterial blood gases, and hemodynamic
variables. An initial PEEP of 5 cm H
2
O is almost always well
tolerated, but blood pressure, heart rate, and pulse oximetry
should be checked immediately before and after the addition.
If the patient is stable, allow 10–20 minutes before arterial
blood gases and cardiac output are determined. If desired
goals are not met, PEEP is increased using selected combina-
tions of PEEP and oxygen concentration. In some patients,
changes in Pa
O
2
may not occur until several hours after PEEP
is changed. These patients cannot be easily identified, but cli-
nicians should be aware that changes in blood gases and
hemodynamics may occur both immediately and long after a
change is made in PEEP.
The goal of PEEP is to facilitate oxygen transfer across the
lungs without impairing systemic oxygen delivery. In most
patients, this is achieved by using the lowest PEEP consistent
with adequate arterial O
2
saturation (>90%). For some time,
the concept of “best PEEP” has been promoted, variously
described as the PEEP level applied to an individual patient
with ARDS that results in the best balance between tissue
oxygenation and adverse effects. Most now agree that the
optimal PEEP is the least that achieves predetermined objec-
tives of patient management rather than some theoretically
ideal value. Thus PEEP should be titrated until Pa
O
2
, arterial
oxygen content, and oxygen delivery increase to acceptable
levels as long as adverse effects are minimized. In practice,
most clinicians prefer PEEP levels between 5 and 12 cm H
2
O
and rarely exceed this range because of fear of barotrauma
and decreased cardiac output. Although respiratory system
PV curves can be determined, most clinicians adjust PEEP
using a combination of response of arterial blood gases,
hypothetical maximum and minimum PEEP values, and
hemodynamic response.
C. Mechanical Ventilation
1. Low-tidal-volume strategy—The most important
development in the management of ARDS is that mechan-
ical ventilation with lower tidal volume than previously
used is associated with improved clinical outcome. This
Step 1: Calculate predicted body weight (PBW) in kg. 0.91 x (height, cm – 152.4) + 50 (for men) or 45.5 (for women).
Step 2: Set ventilatory mode (volume-cycled
assist/control) and tidal volume.
a. Initial tidal volume = 6 mL/kg PBW (if already set higher, then lower 1 mL/kg/h).
b. Measure inspiratory plateau pressure (Pplat) with 0.5 s pause every 4 hours and after every
change in PEEP or tidal volume.
c. Adjust tidal volume based on inspiratory plateau pressure. If Plat >30 cm H
2
O, decrease
tidal volume to 4–5 mL/kg. If Pplat <25 cm H
2
O and tidal volume <6 mL/kg,
increase tidal volume by 1 mL/kg.
Step 3: Adjust respiratory rate. a. Initial respiratory rate to maintain same minute ventilation.
b. Adjust to keep pH 7.30–7.45.
c. Do not exceed rate >35/min or increase rate if Pa
CO
2
<25 mm Hg.
Step 4: Adjust F
IO
2
and PEEP (cm H
2
O) to maintain
Pa
O
2
55–80 mm Hg using only these combinations
Minimize both F
IO
2
and PEEP
F
IO
2
PEEP F
IO
2
PEEP
0.3–0.4 5 0.7 10, 12, 14
0.4 8 0.8 14
0.5 8, 10 0.9 16, 18
0.6 10 1.0 18, 20, 22, 24
Step 5: Manage acidosis or alkalosis as needed. pH <7.30, increase rate (see Step 3)
pH <7.30 and rate = 35, consider bicarbonate administration
pH <7.15, consider increase in tidal volume (even if limited in Step 2)
pH >7.45 and no patient triggering, decrease rate (keep >6/min)
Modified from The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal vol-
umes for acute lung injury and the acute respiratory distress syndrome. N Engl J Med 2000;342:1301–8.
Table 12–20. Lower tidal volume strategy for ARDS.
RESPIRATORY FAILURE
305
has been termed a low-tidal-volume or lung-protective
strategy. In animals, overdistention of lung regions causes
increased epithelial permeability and a histologic picture
of diffuse alveolar damage. In a multicenter study of
ARDS, mortality decreased from 39% (12 mL/kg of pre-
dicted body weight) to 31% in a group whose tidal volume
was set at 6 mL/kg of predicted body weight. Even smaller
tidal volumes were used if inspiratory plateau pressure
remained greater than 30 cm H
2
O. A protocol for adjusting
the mechanical ventilation using a low-tidal-volume strat-
egy is summarized in Table 12–20. Reducing tidal volume
is associated with little or no change in the dead space:tidal
volume ratio and may result in improved O
2
delivery.
Because lung injury is associated with excessive inspiratory
lung volume, higher levels of PEEP may be safer if tidal
volume is limited. Studies have shown little or no adverse
effects of low tidal volume as long as PEEP is given, but
Pa
CO
2
may increase as minute ventilation is decreased with
the lower tidal volume. Low-tidal-volume ventilation with
elevated Pa
CO
2
has been termed permissive hypercapnia.
Acute respiratory acidosis with permissive hypercapnia
was surprisingly well tolerated in several clinical trials.
However, there may be more subtle effects of respiratory
acidosis on nonpulmonary system function, including
renal and neurologic, that are not yet appreciated.
There is not yet agreement on the mechanism of
improved clinical outcome with a low-tidal-volume strategy.
No differences in pneumothorax (explosive barotrauma) are
reported, but there have been small differences in the rate of
nonpulmonary organ dysfunction. An attractive hypothesis
is that ALI is worsened by lung overdistention, and low tidal
volume moderates this “ventilator-associated lung injury.”
For example, the levels of a variety of proinflammatory
cytokines in blood and alveolar fluid are lower in ARDS
patients treated with low tidal volumes. It is likely that the
severe regional heterogeneity of lung involvement is impor-
tant because overdistention would predominate in the most
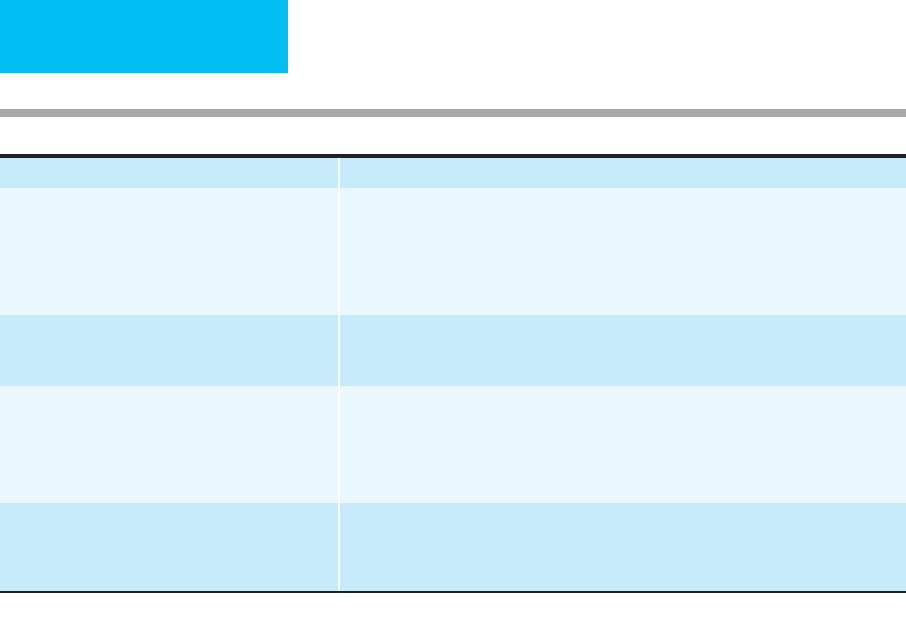
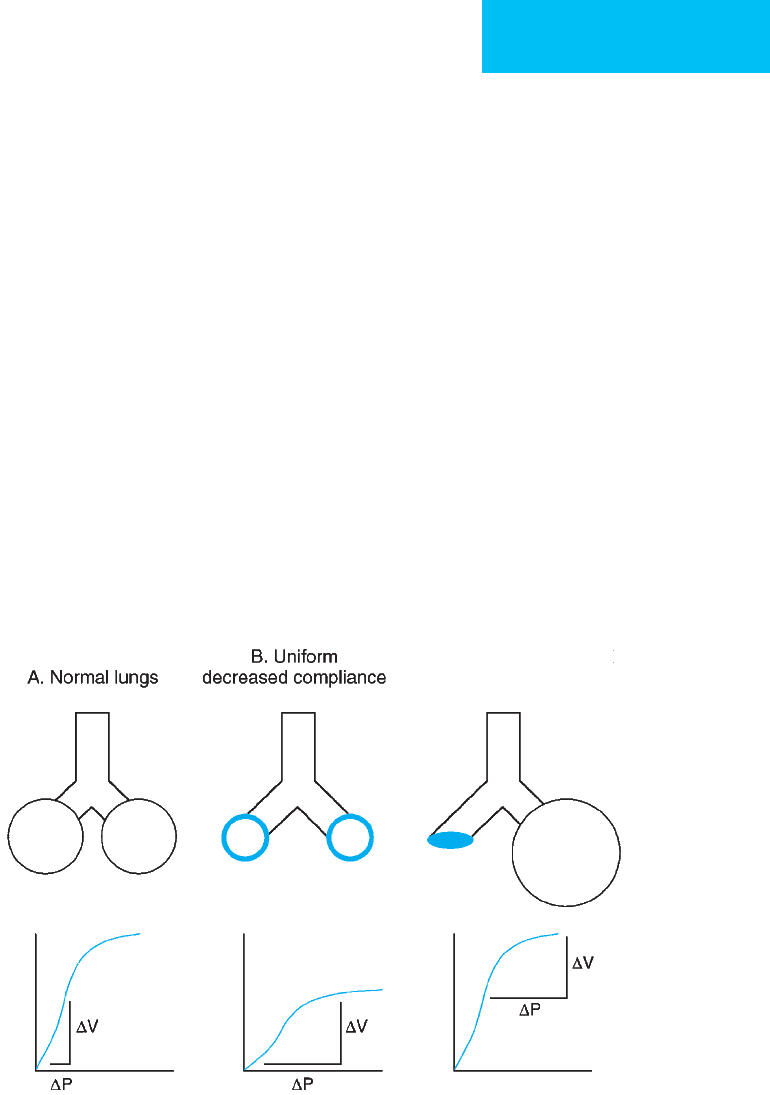
compliant portions of the lungs (Figure 12–10).
Interestingly, there are data supporting the hypothesis that
a tidal volume of 6 mL/kg designed to reach a target of a
plateau pressure of less than 30 cm H
2
O may not be the final
answer. Data suggest that outcome appears to continue to
C. Nonuniform (collapsed
region and normal lung)
Figure 12–10. Schematic illustrating heterogeneity of lung injury in ARDS and effect on respiratory system compli-
ance for the same tidal volume (ΔV) in all three examples.
A.
Normal lungs with uniformly normal compliance. A rep-
resentative PV curve is shown with a small ΔP change as the tidal volume is delivered (ΔV).
B.
Uniformly decreased
compliance of the lungs. The flatter slope of the PV curve results in a larger ΔP required to deliver the same tidal vol-
ume (ΔV).
C.
ARDS is now recognized to have nonuniform lung involvement, and the decreased respiratory system
compliance arises from collapse of the majority of lung units and overdistention of remaining normal units. The same
tidal volume is delivered to a smaller region of normal lung so that ΔP is large for the same ΔV. The overall compli-
ance shown in
C
(ΔV/ΔP) is the same as in
B
.

CHAPTER 12
306
improve as tidal volume or targeted plateau pressure
decreases. This likely would mean that some patients would
benefit from a tidal volume of less than 6 mL/kg, although
not all would require this. Further randomized trials are
needed.
2. Volume-preset ventilation—Most ARDS patients are
ventilated using conventional volume-preset (volume-
cycled) positive-pressure ventilators. The major studies using
low tidal volume for ARDS used this mode. Initial tidal vol-
ume is set at 6 mL/kg of ideal body weight, and peak inspira-
tory flow rate is generally at least 1–1.2 L/s owing to the high
demand for inspiratory flow. Adjustments in tidal volume
depend on the level of the inspiratory plateau pressure (P
plat
),
as shown in Table 12–20. PEEP is given as necessary for
refractory hypoxemia and in order to lower the F
IO
2
using a
set of predetermined values for each variable.
Volume-preset ventilation using a high peak flow and a
descending inspiratory flow pattern may have characteristics
similar to those of pressure-controlled ventilation (PCV). In
theory, these settings may improve distribution of ventila-
tion to poorly ventilation lung regions, improving hypox-
emia with smaller increases in PEEP or F
IO
2
.
3. Pressure-controlled ventilation—Pressure-controlled
ventilation (PCV) might seem like a very attractive option in
the management of ARDS, largely because a preset maxi-
mum positive airway pressure cannot be exceeded. However,
this feature does not automatically provide the same benefit
as a low-tidal-volume strategy unless the maximum pressure
is adjusted to also limit tidal volume. On the other hand,
PCV does have the theoretical advantage of providing maxi-
mum inspiratory flow at the beginning of the inspired
breath. In some patients, distribution of ventilation may be
enhanced, especially to the most poorly ventilated lung
regions.
A few studies have shown that PCV compared with con-
ventional volume-preset ventilation results in increased
Pa
O
2
, decreased mean and peak airway pressures, and less
impairment of cardiac output. Other studies have shown no
appreciable differences. Careful adjustment of inspiratory
time is needed to optimize tidal volume and minute ventila-
tion. PCV with pressure limited to 30–40 cm H
2
O might be
considered in ARDS patients with severe hypoxemia unre-
sponsive to PEEP with conventional volume-preset ventila-
tion or in those who require excessively high airway pressures
or PEEP with conventional ventilation.
Similarly, airway pressure-release ventilation (APRV; see
above) has some attractive features for ventilatory support in
ARDS. Lung recruitment might be improved because of the
higher mean airway pressure, and there may be reduced
stress-relaxation lung injury. No studies, however, have
demonstrated improved outcome in ARDS with APRV com-
pared with more conventional modes. For refractory hypox-
emia, a trial of APRV might be useful and could be used in
comparison to IRV, prone ventilation, and maximum-lung-
recruitment strategies.
4. Inverse-ratio ventilation—In ARDS patients with
refractory hypoxemia, inverse-ratio ventilation (IRV) has
been used. In contrast to conventional ventilation, inspira-
tory time is made longer than expiratory time by decreasing
the inspiratory flow rate, holding inspiration for a preset
time before allowing for exhalation, or, if a time-cycled ven-
tilator is used, directly increasing inspiratory time. How IRV
might improve oxygenation is not clear. Inspired gas may be
distributed more evenly because of the longer inspiratory
time. The shortened expiratory time, on the other hand, may
cause dynamic hyperinflation, raising end-expiratory vol-
ume and improving gas exchange in a manner similar to
intrinsic PEEP.
There have been no controlled clinical trials demonstrat-
ing improved outcome with IRV. This method should be
reserved for the rare patient with refractory hypoxemia unre-
sponsive to PEEP and oxygen therapy. Current understand-
ing of IRV suggests that there is nothing intrinsically
different about a I:E ratio greater than 1:1. Rather, the gas-
exchange effects of I:E ratio vary continually.
When initiated, an I:E ratio of 1:1 should be tried, and
blood pressure, heart rate, and pulse oximetry should be
monitored closely. If needed, I:E ratio can be further altered
to 1.5:1, 2:1, or more. It is unusual for I:E ratios of greater
than 3:1 or 4:1 to improve gas exchange.
Increased time spent during inspiration and dynamic
hyperinflation can cause severely reduced cardiac output and
hypotension. Prolonged inspiratory time may be very
uncomfortable to the patient, and sedation or muscle relax-
ants are always needed. Monitoring of IRV is complex
because I:E ratio, peak airway pressure, and PEEP do not
adequately reflect all the essential parameters and because
the pattern of inspiratory pressure and flow in IRV differs
depending on how IRV is produced. Monitoring mean air-
way pressure has been suggested, but this value does not
correlate with gas exchange or hemodynamic compromise
in IRV.
5. Other modes of mechanical ventilation—Lung-
protective strategies are the basis for several other modes of
mechanical ventilation. Carrying low tidal volume to
extreme is extracorporeal membrane oxygenation and CO
2
removal, in which the lungs receive no ventilation at all.
Clinical trials have been unable to demonstrate improved
clinical outcomes from this practice. High-frequency oscilla-
tion has been used for more than 20 years. With this method,
very small tidal volumes (1–2 mL/kg or less) at respiratory
rates as high as several hundred per minute are administered
by an oscillating membrane. High-frequency oscillation has
been successful in neonatal respiratory distress, but scaling
up to larger patients has not been uniformly feasible. Gas
exchange often can be maintained or improved, but no dif-
ference in clinical outcome has been demonstrated. Another
lung-protective method is tracheal gas insufflation, in which
a 4–6 L/min flow of fresh gas is provided into a small catheter
placed in the lower trachea. The effect is to reduce the apparent