Voet D., Voet Ju.G. Biochemistry
Подождите немного. Документ загружается.

GTP (or the 10-fold less abundant GDP) can subsequently
displace Sos from Ras.
The different families of small G proteins interact with
different classes of GEFs, whose catalytic domains share
no sequence similarity and are structurally unrelated [e.g.,
rhodopsin (Fig. 19-16) is a GEF for G
t␣
]. Nevertheless,
many of these GEFs share the same general mechanism for
promoting GDP–GTP exchange, which suggests that this
mechanism arose on several occasions through convergent
evolution.
i. GAPs Function to Turn Off Ras-Mediated Signals
Ras hydrolyzes its bound GTP with a rate constant of
0.02 min
⫺1
(vs 2–3 min
⫺1
for G
␣
subunits), too slowly for ef-
fective signal transduction. This led to the discovery of a
120-kD GTPase activating protein, RasGAP, that, on bind-
ing Ras ⴢ GTP, accelerates the rate of GTP hydrolysis by a
factor of 10
5
. RasGAP’s physiological importance as a
regulator of Ras-mediated signal transduction is demon-
strated by the observation that the relative biological
activities of Ras mutants are better correlated with their
resistance to regulation by RasGAP than by their intrinsic
GTPase activity.
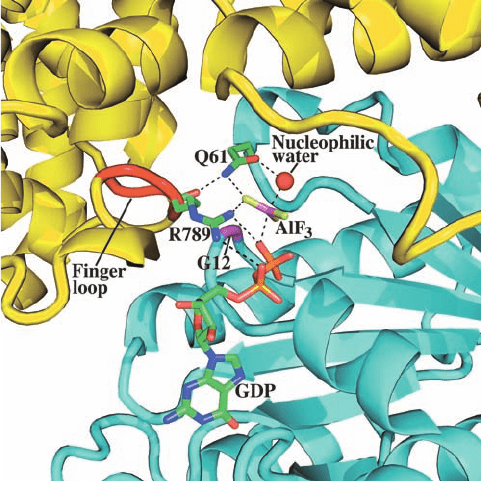
The mechanism whereby RasGAP activates the GTPase
activity of Ras was revealed by the X-ray structure, deter-
mined by Alfred Wittinghofer, of the 334-residue GTPase-
activating domain of RasGAP (GAP334) bound to Ras in
its complex with GDP and AlF
3
(Fig. 19-39). GAP334,
which consists of two all-helical domains, interacts with
Ras over an extensive surface that includes its Switch I and
II regions. The AlF
3
, which has trigonal planar symmetry,
binds to Ras at the expected position of GTP’s ␥ phosphate
group, with the Al atom opposite a bound water molecule
that presumably would be the attacking nucleophile in the
GTPase reaction. Since Al¬F and P¬O bonds have simi-
lar lengths and phosphoryl-transfer reactions occur via a
trigonal bipyramidal transition state (Fig. 16-6b), the
GDP–AlF
3
–H
2
O assembly presumably resembles the
GTPase reaction’s transition state, with the AlF
3
mimicking
the planar PO
3
group. Note that Ras ⴢ GDP does not by
itself bind AlF
3
.
GAP334 binds to Ras with GAP334’s exposed so-called
finger loop inserted into the Ras active site such that the
finger loop’s Arg 789 side chain interacts with both the
Ras-bound GDP’s  phosphate and the AlF
3
(Fig. 19-39).
In Ras ⴢ GTP, this Arg side chain would be in an excellent
position to stabilize the developing negative charge in the
GTPase reaction’s transition state. Indeed, the catalytically
more efficient G
␣
subunits contain an Arg residue (Arg 178
in G
i␣
), whose guanidinium group occupies a nearly identi-
cal position (in G
s␣
, this is the Arg side chain that is ADP-
ribosylated by cholera toxin; Section 19-2Cd). The main
chain carbonyl O of Arg 789 hydrogen bonds to the side
chain N of Ras’ catalytically important Gln 61. The O of
this side chain is thereby positioned to hydrogen bond with
the nucleophilic water molecule while its NH
2
group inter-
acts with an F atom of AlF
3
(Fig. 19-37), an arrangement
that presumably stabilizes the GTPase reaction’s transition
state.
j. Oncogenic Mutants of Ras Are GAP-Insensitive
Mutations in Ras of Gly 12 and Gln 61 are its most com-
mon oncogenic mutations (an oncogenic form of which is
found in ⬃30% of human cancers). These mutations pre-
vent RasGAP from activating Ras to hydrolyze its bound
GTP and hence lock Ras into its active conformation. The
foregoing X-ray structure reveals why these mutants are
GAP-insensitive. Gly 12 is in such close proximity to the
finger loop that even the smallest possible residue change
(to Ala) would sterically interfere with the geometry of the
transition state through steric clashes with the main chain
of Arg 789 (of RasGAP) and the side chain NH
2
of Gln 61.
The observation that Gly 12 mutants of Ras bind GTP with
nearly wild-type affinity therefore suggests that larger
side chains can be tolerated at Ras residue 12 in the
Ras–RasGAP Michaelis complex but not in the transition
state. The apparent participation of Gln 61 in transition
state stabilization confirms that this residue has an essen-
tial role in catalysis.
Section 19-3. Tyrosine Kinase–Based Signaling 711
Figure 19-39 X-ray structure of the GAP334 ⴢ Ras ⴢ GDP ⴢ
AlF
3
complex. The active site regions of the proteins are
shown as ribbons with Ras cyan, its Gly 12 magenta, GAP334
yellow, and its finger loop red.The GDP, AlF
3
, and the side
chains of Ras Asn 61 and GAP334 Arg 789 are drawn in stick
form with C green, N blue, O red, F yellow-green, P orange, and
Al pink; the nucleophilic water molecule is represented by a red
sphere; and hydrogen bonds are shown as dashed lines. [Based
on an X-ray structure by Alfred Wittinghofer, Max-Planck-Institut
für Molekulare Physiologie, Dortmund, Germany. PDBid
1WQ1.]
JWCL281_c19_671-743.qxd 7/1/10 1:12 PM Page 711
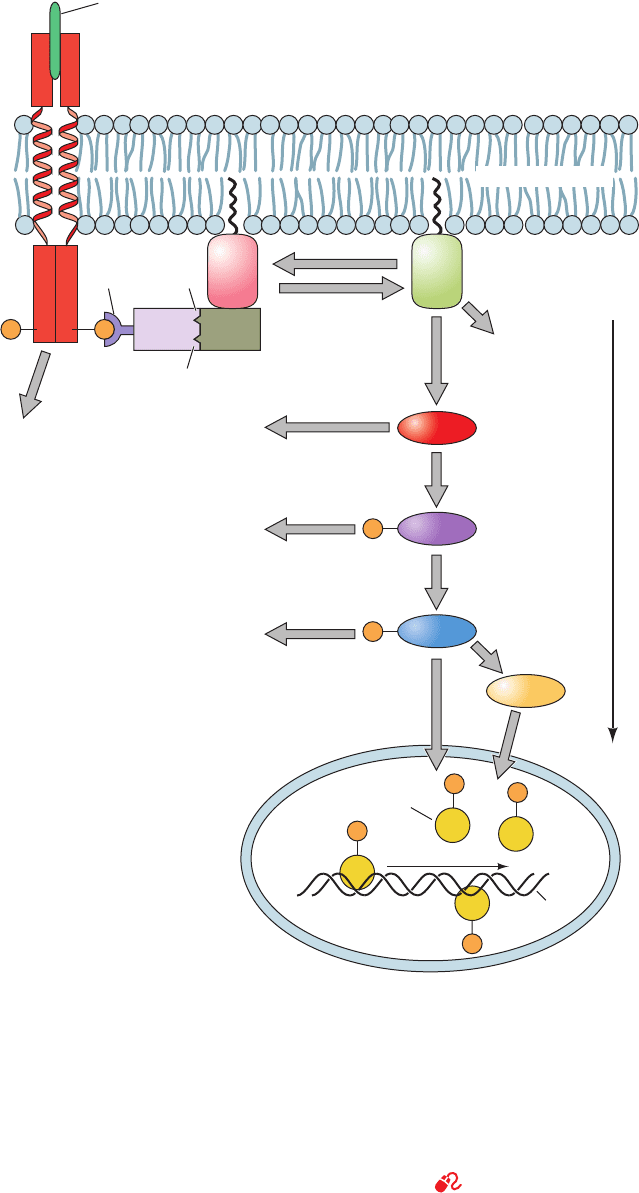
D. MAP Kinase Signaling Cascades
The signaling pathway downstream of Ras consists of a
linear series of Ser/Thr kinases that form a so-called MAP
kinase cascade (Fig. 19-40). Many of the proteins that par-
ticipate in MAP kinase cascades are the products of proto-
oncogenes:
1. Raf, a Ser/Thr protein kinase, is activated by direct
interaction with Ras ⴢ GTP (although other signaling
712 Chapter 19. Signal Transduction
Figure 19-40 The Ras-activated MAP kinase cascade. This
signaling cascade begins when an RTK binds its cognate growth
factor, thereby inducing the autophosphorylation of this RTK’s
cytosolic domain. Grb2/Sem-5 binds to the resulting
phosphoTyr-containing peptide segment via its SH2 domain
and simultaneously binds to Pro-rich segments on Sos via its two
SH3 domains.This activates Sos as a guanine nucleotide
exchange factor (GEF) to exchange Ras’ bound GDP for GTP,
which activates Ras to bind to Raf. Then, Raf, a Ser/Thr kinase,
Myc
Transcription
factors
Gene expression
Nucleus
DNA
Jun
P PYY
P
P
P
Sos
Other effectors
Other
effectors
Other effectors
Other effectors
Sos
Other
effectors
(GAPs)
GAPs Cytosol
Extracellular
medium
Kinase
cascade
Grb2/
Sem–5
SH3
domain
SH3
domain
SH2
domain
Protein
growth
factor
RTK
Raf
MEK
MAPK
Other
kinases
Fos
P
Jun
P
P
Plasma membrane
Ras
(inactive)
GTP
Ras
(active)
GDP
phosphorylates MEK, which in turn phosphorylates MAPK,
which then migrates to the nucleus, where it phosphorylates
transcription factors such as Fos, Jun, and Myc, thereby
modulating gene expression.The MAP kinase cascade eventually
returns to its resting state through the actions of protein
phosphatases (Section 19-3F) after a GTPase activating protein
(GAP) deactivates Ras by inducing it to hydrolyze its bound
GTP to GDP. [After Egan, S.E. and Weinberg, R.A., Nature 365,
782 (1993).]
See the Animated Figures
JWCL281_c19_671-743.qxd 6/4/10 10:54 AM Page 712
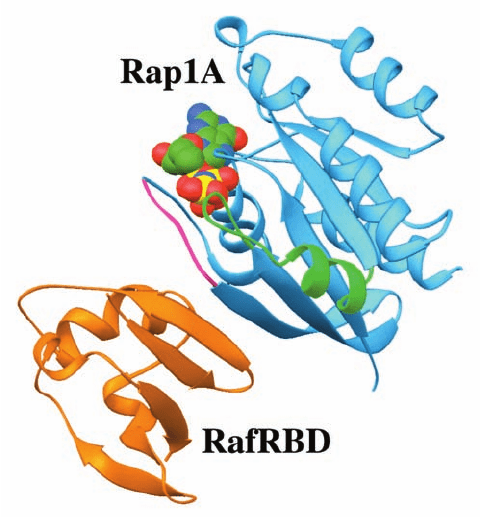
pathways may activate Raf by phosphorylating it at multi-
ple Ser and Thr residues;see below).The X-ray structure of
the Ras homolog Rap1A in complex with GMPPNP and
the Ras binding domain of Raf (RafRBD), determined by
Wittinghofer, reveals that the two proteins associate
largely by mutually extending their antiparallel sheets
across a mainly polar interface (Fig. 19-41). Although
Ras GTP has 1300-fold greater affinity for binding to Raf
than does Ras GDP, it is unclear from this structure how
GTP hydrolysis by Ras affects the Ras–Raf interface. Quite
possibly the conformational change in Ras Switch I per-
turbs the Ras–Raf interface to the point that it dissociates.
2. Activated Raf phosphorylates a protein alternatively
known as MEK and MAP kinase kinase (MKK) at specific
Ser and Thr residues, thereby activating it as a Ser/Thr kinase.
[Raf is therefore a MAP kinase kinase kinase (MKKK)].
3. Activated MEK phosphorylates a family of proteins
named mitogen-activated protein kinases (MAP kinases or
MAPKs) or extracellular-signal-regulated kinases (ERKs).
For more than marginal activation, a MAPK must be phos-
phorylated at both its Thr and Tyr residues in the sequence
Thr-X-Tyr. MEK (which stands for MAP kinase/ERK-
activating kinase) catalyzes both phosphorylations and
thus has dual specificity for Ser/Thr and Tyr. The X-ray
structure of the unphosphorylated MAP kinase ERK2, de-
termined by Elizabeth Goldsmith, reveals that this protein
structurally resembles other protein kinases of known
structure and that its Tyr residue that becomes phosphoryl-
ated blocks the peptide-binding site in its unphosphoryl-
ated form.
4. The activated MAPKs phosphorylate a variety of cy-
toplasmic and membrane-associated proteins, including
Sos and EGFR, at Ser/Thr-Pro motifs. In addition, the
MAPKs migrate from the cytosol to the nucleus, where
they phosphorylate a large variety of transcription factors
including Jun/AP-1, Fos, and Myc. These activated tran-
scription factors, in turn, induce the transcription of their
target genes (Section 34-4Bd). The effects commissioned
by the extracellular presence of the protein growth factor
that initiated the signaling cascade are thereby produced.
MAP kinase cascades can be activated in other ways be-
sides by liganded RTKs. For example, Raf may also be acti-
vated via its Ser/Thr phosphorylation by protein kinase C,
which is activated via the phosphoinositide signaling sys-
tem described in Section 19-4. Alternatively, Ras may be
activated by subunits of certain heterotrimeric G proteins.
Thus, the MAP kinase cascade serves to integrate a variety
of extracellular signals.
a. Scaffold and Anchoring Proteins Organize
and Position Protein Kinases
Eukaryotic cells contain numerous different MAPK
signaling cascades, each with a characteristic set of com-
ponent kinases, which in mammals comprise at least 14
MKKKs, 7 MKKs, and 12 MAPKs (Fig. 19-42). Although
each MAPK is activated by a specific MKK, a given MKK
can be activated by more than one MKKK. Moreover, sev-
eral pathways may be activated by a single type of recep-
tor. How then does a cell prevent inappropriate cross talk
between closely related signaling pathways? One way that
this occurs is through the use of scaffold proteins, proteins
that bind some or all of the component protein kinases of a
particular signaling cascade so as to ensure that the protein
kinases of a given pathway interact only with one another.
In addition, a scaffold protein can control the subcellular
location of its associated kinases.
The first known scaffold protein was discovered through
the genetic analysis of a MAP kinase cascade in yeast,
which demonstrated that this protein, Ste5p, binds the
MKKK, MKK, and MAPK components of the pathway
and that, in vivo, the scaffold’s absence inactivates the
pathway. Evidently, the interactions between successive
kinase components of this MAP kinase cascade are, by
themselves, insufficient for signal transmission.
Section 19-3. Tyrosine Kinase–Based Signaling 713
Figure 19-41 X-ray structure of the Ras binding domain of
Raf (RafRBD; orange) in complex with Rap1A GMPPNP
(blue). The Switch I and II regions of Rap1A are magenta and
green, and its bound GMPPNP is shown in space-filling form
with C green, N blue, O red, and P yellow. Rap1A GMPPNP
and Ras GMPPNP have nearly identical structures. [Based on
an X-ray structure by Alfred Wittinghofer, Max-Planck-Institut
für Molekulare Physiologie, Dortmund, Germany. PDBid
1GUA.]
JWCL281_c19_671-743.qxd 3/16/10 7:17 PM Page 713
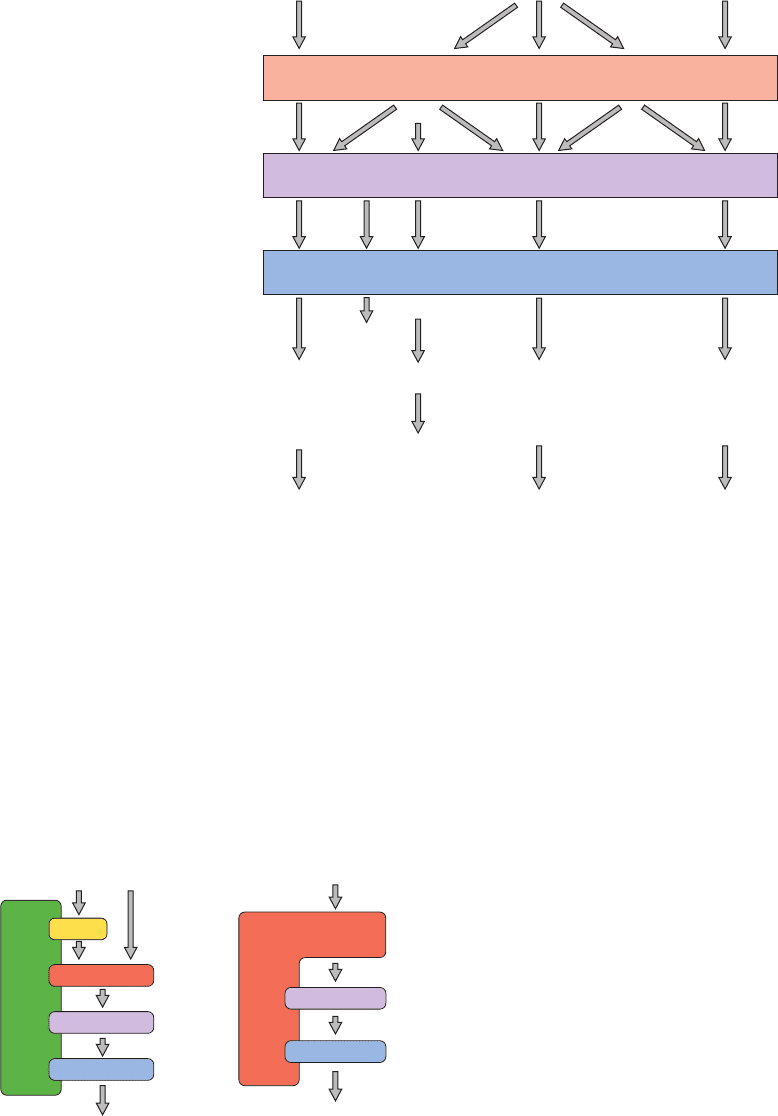
JIP-1 (for JNK interacting protein-1; Fig. 19-43a) is a
scaffold protein that simultaneously binds HPK1 (for
hematopoietic progenitor kinase-1), a Ras analog and
hence an MKKK kinase (MKKKK); the MKKKs MLK3
and DL3; MKK7; and the MAPK JNK (for Jun N-terminal
kinase). MEKK1 is a somewhat different type of scaffold
protein (Fig. 19-43b); this functional MKKK binds its sub-
strate, MKK4, as well as the latter’s substrate, JNK.
Protein Ser/Thr kinases may similarly be individually
tethered to their sites of action by anchoring proteins. For
example, protein kinase A (PKA), which participates in nu-
merous parallel signaling pathways including that regulat-
ing glycogen metabolism (Section 18-3), associates with
several unrelated so-called A-kinase anchoring proteins
(AKAPs). The different AKAPs, which all bind the re-
gulatory (R) subunits of PKA, target PKA to different
subcellular locations (e.g., to vesicle or plasma membranes
or to particular receptors) and may also bind other signaling
proteins (e.g., PP1, the protein phosphatase that removes
phosphate groups installed by PKA; Section 18-3Cg), thus
functioning to integrate intracellular signals.
b. Anthrax Lethal Factor Specifically
Cleaves MAPKKs
Anthrax, an infectious disease caused by the bacterium
Bacillus anthracis, affects mainly herbivorous animals such
as cattle, sheep, and goats. On rare occasions, however, it
may be transmitted to humans (but not between humans),
in whom it is often fatal, if untreated, through massive
714 Chapter 19. Signal Transduction
Figure 19-43 Some examples of scaffold proteins that
modulate mammalian MAP kinase cascades. (a) JIP-1 binds all
protein components of the MAP kinase cascade in which HPK1
phosphorylates MKL3 or DLK (MKKKs), which then
phosphorylates MKK7, which then phosphorylates JNK (an
MAPK). (b) MEKK1 (an MKKK) is the kinase for MKK4 and
also binds JNK, MKK4’s target MAPK. [After Garrington,T.P.
and Johnson, G.L., Curr. Opin. Cell Biol. 11, 213 (1999).]
Figure 19-42 MAP kinase cascades in mammalian cells. Each
MAP kinase cascade consists of an MKKK, an MKK, and an
MAPK.Various external stimuli may each activate one or more
MKKKs, which in turn may activate one or more MKKs.
However, the MKKs are relatively specific for their target
MAPKs.The activated MAPKs phosphorylate specific
Growth factor
Extracellular
stimulus:
Stress, growth factor,
differentiation factor
Transcription
factors and
other kinases:
Cellular
response:
MEKK4,
DLK
TAK1, ASK1,
MLK3
PAKMKKK
MEKK1-3,
Tpl-2
Raf-1, A-Raf,
B-Raf, Mos
MKK3, MKK6MKK4, MKK7MKK5
?
?
?
MKK
MEK1,
MEK2
JNK1, JNK2,
JNK3
p38α, p38β,
p38γ, p38δ
ERK5
MEF2C
c-Jun
MAPK
ERK3,
ERK4
ERK1,
ERK2
c-Jun, ATF-2, Elk-1, p53,
DPC4, NFAT4
MAPKAP kinase, ATF-2,
Elk-1, Chop, Max, MEF2C
p90
rsk
, S6 kinase, Sos,
phospholipase A
2
,
EGF receptor, Elk-1, Ets1,
Sap1a, c-Myc, Tal, STATS
Growth, differentiation,
survival, apoptosis
Cytokine production,
apoptosis
Growth,
differentiation
Stress
MLK3 or DLK
Cytokines, stress, etc. Stress, etc.
HPK1
MKK7
JNK
Response
Response
JNK
MKK4
MEKK1JIP1
(a) (b)
transcription factors (e.g., Elk-1, Ets1, p53, NFAT4, Max) as well
as specific kinases (e.g., p90
rsk
, S6 kinase, MAPKAP kinase). The
resulting activated transcription factors and kinases then induce
cellular responses such as growth, differentiation, and apoptosis
(programmed cell death; Section 34-4E). [After Garrington, T.P.
and Johnson, G.L., Curr. Opin. Cell Biol. 11, 212 (1999).]
JWCL281_c19_671-743.qxd 3/16/10 7:17 PM Page 714
septic shock (Section 19-1Lb). Anthrax spores are signifi-
cant agents of biological warfare because their inhalation
results in inhalational anthrax, a form of the disease that is
nearly always fatal. This is because by the time the symp-
toms of inhalational anthrax become apparent,the bacterial
infection has already released so much toxin that eliminat-
ing the infection through antibiotic treatment does not
reverse the progress of the disease.
Anthrax toxin consists of three proteins that act in con-
cert: protective antigen (PA), lethal factor (LF), and
edema factor (EF; oedema factor in British English). PA,
which is named for its use in vaccines, is a 735-residue, 4-
domain protein that binds to its host cell-surface receptor
(a single pass transmembrane protein) via its C-terminal
domain. Most of PA’s N-terminal domain is then cleaved
away by a cell-surface protease, whereon the remaining
membrane-bound portions of PA form cyclic heptamers
reminiscent of the cyclic pentamers formed by cholera
toxin (Fig. 19-22).The heptameric PA then binds LF and/or
EF by their homologous N-terminal domains and mediates
their endocytotic uptake into the cell. Indeed, the intra-
venous administration of only PA and LF rapidly kills ani-
mals. EF is a calmodulin-activated adenylate cyclase whose
action upsets water homeostasis and hence is probably re-
sponsible for the massive edema (abnormal buildup of in-
tercellular fluids) seen in cutaneous anthrax infections.
LF is a 776-residue monomeric protease that has only
one known cellular target: It cleaves members of the
MAAPKK family of proteins near their N-termini so as to
excise the docking sequences for their cognate downstream
MAPKs. It thereby disrupts the signal transduction path-
ways in which these proteins participate. However, anthrax
infection targets mainly macrophages, a type of white
blood cell (mice whose blood has been depleted of
macrophages are resistant to anthrax). Low levels of LF,
which occur in early stages of anthrax infection, cleave
MAPKK-3, which inhibits macrophages from releasing but
not producing the inflammatory mediators NO (Section
19-1Lb) and tumor necrosis factor- (TNF; a cytokine
that has opposite effects to most protein growth factors
and is largely responsible for the wasting seen in chronic
infections). This has the effect of reducing and/or delaying
the immune response. In contrast, high levels of LF, which
occur in late stages of infection, trigger macrophage lysis,
causing the sudden release of NO and TNF-, which pre-
sumably results in the massive septic shock that causes
death.
E. Tyrosine Kinase–Associated Receptors
Many cell-surface receptors are not members of the recep-
tor families that we have discussed and do not respond to
ligand binding by autophosphorylation. These include the
receptors for the cytokines (Section 19-3Eb) and T cell re-
ceptors [which control the proliferation of immune system
cells known as T lymphocytes (T cells); Section 35-2D].
Ligand binding induces these tyrosine kinase–associated
receptors to dimerize (and, in some cases, to trimerize or
form even higher oligomers), often with different types of
subunits, in a way that activates associated nonreceptor ty-
rosine kinases (NRTKs). The domain organization for the
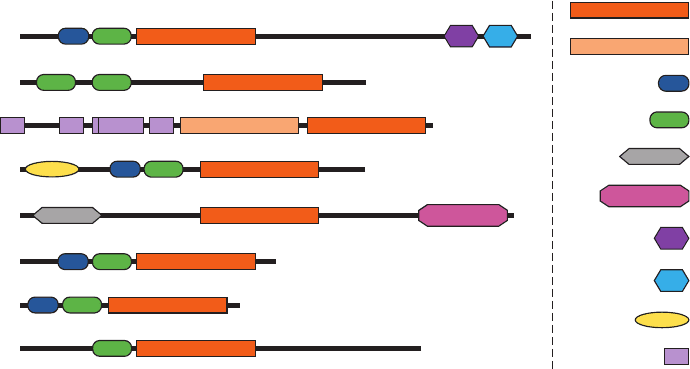
major subfamilies of NRTKs is diagrammed in Fig. 19-44.
a. The Structure of Src Reveals Its
Autoinhibitory Mechanism
Many of the NRTKs that are activated by tyrosine
kinase–associated receptors belong to the Src family, which
contains at least nine members including Src, Fyn, and
Lck. Most of these ⬃530-residue membrane-anchored (by
myristoylation) proteins have both an SH2 and an SH3
domain and all have a PTK domain. Hence, an Src-related
kinase may also be activated by association with an au-
tophosphorylated RTK. Although Src-related kinases are
each associated with different receptors, they phosphorylate
Section 19-3. Tyrosine Kinase–Based Signaling 715
Figure 19-44 Domain organization of the major NRTK
subfamilies. The N-termini for these polypeptides, which are
drawn approximately to scale, are on the left and the domain
Abl
tyrosine kinase
pseudo-kinase
SH3
SH2
integrin binding
focal adhesion binding
DNA binding
F-actin binding
pleckstrin homology
Jak homology
Zap70
Jak
Btk
Fak
Src
Csk
Fes
identification is provided on the right. [Courtesy of Stevan
Hubbard, New York University School of Medicine.]
JWCL281_c19_671-743.qxd 3/16/10 7:17 PM Page 715
overlapping sets of target proteins. This complex web of
interactions explains why different ligands often activate
some of the same signaling pathways.
Src, as indicated in Fig. 19-44, consists of, from N- to
C-terminus, a myristoylated N-terminal “unique” domain
that differs among Src family members, an SH3 domain, an
SH2 domain, a PTK domain, and a short C-terminal tail.
Phosphorylation of Tyr 416 in the PTK’s activation loop
activates Src, whereas phosphorylation of Tyr 527 in its
C-terminal tail deactivates it. In vivo, Src is phosphorylated
at either Tyr 416 or Tyr 527, but not at both. The dephos-
phorylation of Tyr 527 or the binding of external ligands to
the SH2 or the SH3 domain activates Src, a state that is
then maintained by the autophosphorylation of Tyr 416.
When Tyr 527 is phosphorylated and no activating phos-
phopeptides are available, Src’s SH2 and SH3 domains
function to deactivate its PTK domain, that is, Src is then
autoinhibited.
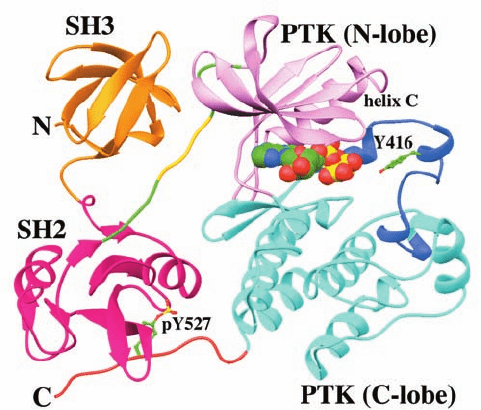
The X-ray structure of Src AMPPNP lacking its
N-terminal domain and with Tyr 527 phosphorylated, deter-
mined by Stephen Harrison and Michael Eck, reveals the
structural basis of Src autoinhibition (Fig. 19-45). As bio-
chemical studies had previously shown, the SH2 domain
binds phosphoTyr 527, which occurs in the sequence
pYNPG rather than the pYEEI sequence characteristic of
high affinity Src SH2 target peptides. Although the pYNP
segment binds to SH2 as does the pYEE segment in Fig.
19-33b, the succeeding residues are poorly ordered in the
X-ray structure and, moreover, the SH2 pocket in which
the Ile side chain of pYEEI binds is unoccupied. Appar-
ently, the phosphoTyr 527-containing peptide segment
binds to the Src SH2 domain with reduced affinity relative
to its target peptides.
The SH3 domain binds to the linker connecting the SH2
domain to the N-terminal lobe of the PTK domain.
Residues 249 to 253 of this linker form a polyproline II he-
lix that binds to the SH3 domain in much the same way as
do SH3’s Pro-rich target peptides (Fig. 19-33). However,
the only Pro in this segment is residue 250. The polar side
chain of Gln 253, which occupies the position of the second
Pro in SH3’s normal Pro-X-X-Pro target sequence, does
not enter the hydrophobic binding pocket that this second
Pro would occupy (Fig. 19-35), and hence the path of the
peptide deviates from that of Pro-rich target peptides at
this point. Apparently, this interaction is also weaker than
those with Src’s SH3 target peptides.
Src’s SH2 and SH3 domains bind on the opposite side of
the PTK domain from its active site. How, then, does the
conformation shown in Fig.19-45 inhibit the PTK’s activity?
The two lobes of Src’s PTK domain are, for the most part,
closely superimposable on their counterparts in the PTK
domain of phosphorylated and hence activated Lck (a Src
family member) as well as the C subunit of activated PKA
(Fig. 18-15). However, Src helix C (the only helix in the
PTK’s N-terminal lobe) is displaced from the interface be-
tween the N- and C-terminal lobes relative to its counter-
parts in Lck and PKA. Helix C contains the conserved
residue Glu 310 (using Src numbering), which in activated
Lck and PKA projects into the catalytic cleft,where it forms
a salt bridge with Lys 295, an important ligand of the sub-
strate ATP’s and phosphates. However, in inactive Src,
Glu 310 forms an alternative salt bridge with Arg 409,
whereas Lys 295 instead interacts with Asp 404. In activated
Lck,Arg 409 forms a salt bridge with phosphoTyr 416.
The foregoing structural observations suggest the fol-
lowing scenario for Src activation (Fig. 19-46):
1. The dephosphorylation of Tyr 527 and/or the binding
of the SH2 and/or SH3 domains to their target peptides
(for which SH2 and SH3 have greater affinity than their in-
ternal Src binding sites) releases these domains from their
PTK-bound positions shown in Fig. 19-45, thus relaxing
conformational constraints on the PTK domain. This al-
lows the PTK’s active site cleft to open, thereby disrupting
the structure of its partially helical activation loop (which
occupies a blocking position in the active site cleft; Fig.
19-45) so as to expose Tyr 416 to autophosphorylation.
2. The resulting phosphoTyr 416 forms a salt bridge
with Arg 409, which sterically requires the structural reor-
ganization of the activation loop to its active, nonblocking
conformation.The consequent rupture of the Glu 310–Arg
409 salt bridge frees helix C to assume its active orientation
which, in turn, allows Glu 310 to form its catalytically im-
portant salt bridge to Lys 295, thereby activating the Src
PTK activity.
716 Chapter 19. Signal Transduction
Figure 19-45 X-ray structure of Src AMPPNP lacking its
N-terminal domain and with Tyr 527 phosphorylated. The SH3
domain is orange, the SH2 domain is magenta, the linker joining
the SH2 domain to the PTK domain is green with its 5-residue
polyproline II helix gold, the N-terminal lobe of the PTK domain
is pink, the C-terminal lobe is cyan with its activation loop blue,
and the C-terminal tail is orange. The AMPPNP is shown in
space-filling form and Y416 and pY527 are shown in ball-and-stick
form, all with C green, N blue, O red, and P yellow. [Based on an
X-ray structure by Stephen Harrison and Michael Eck, Harvard
Medical School. PDBid 2SRC.]
JWCL281_c19_671-743.qxd 3/16/10 7:17 PM Page 716

The above mechanism is, perhaps unexpectedly, criti-
cally dependent on the rigidity of the 8-residue linker join-
ing the SH2 and SH3 domains. Thus, replacing three of
these linker residues with Gly (whose lack of a C
atom
makes it the least conformationally restricted residue) re-
sults in a protein that is no longer deactivated by the phos-
phorylation of Tyr 527. This is corroborated by molecular
dynamics simulations (Section 9-4) indicating that the ther-
mal motions of the SH2 and SH3 domains are highly corre-
lated (move as a unit) when Tyr 527 is phosphorylated but
that this correlation is significantly reduced when Tyr 527 is
dephosphorylated or when Gly replaces the three linker
residues.
b. The JAK-STAT Pathway Relays
Cytokine-Based Signals
Cytokines form a diverse group of small soluble pro-
teins that, when secreted by cells, act in an autocrine,
paracrine, or endocrine fashion to induce a great variety of
responses, including the immune response (Section 35-2Aa),
cell proliferation, growth, differentiation, apoptosis (pro-
grammed cell death; Section 34-4E), and chemotaxis (the
movement of a motile cell along a concentration gradient
of a specific substance).They include the 35 different inter-
leukins (IL-1 to IL-35), the colony stimulating factors [which
include macrophage colony-stimulating factor (M-CSF),
granulocyte colony-stimulating factor (G-CSF), and granu-
locyte macrophage colony-stimulating factor (GM-CSF);
macrophages and granulocytes are types of white blood
cells], growth hormone (Section 19-1J), erythropoietin
(EPO; which stimulates the production of erythrocytes),
the interferons (which protect against viral infection; Sec-
tion 32-4Ab), the tumor necrosis factors (TNFs), nerve
growth factor (NGF), and the chemokines (which induce
chemotaxis in responsive cells). Abnormalities in specific
cytokines or their receptors have been implicated in a large
variety of diseases and, conversely, several cytokines are
therapeutically useful in alleviating certain pathological
states [e.g., EPO is used to treat anemia resulting from
chronic kidney disease (EPO is produced by the kidneys)
and GM-CSF is used to stimulate white cell production fol-
lowing chemotherapy (which kills fast-growing cells such
as white cells)].
Most cytokine receptors, as we saw for human growth
hormone (Section 19-1J), are activated through ligand-
induced receptor aggregation of two or more receptor
components. For example, the GM-CSF receptor consists
of two different types of subunits: GMR, which is specific
for GM-CSF, and
c
, which is a common subunit for the
GM-CSF, IL-3, and IL-5 receptors. Both of these subunits
consist of an N-terminal cytosolic domain, a single trans-
membrane helix, and a C-terminal ectodomain, which in
GMR consists of two fibronectin type III domains (Sec-
tion 19-1J) and in
c
consists of four such domains.
The X-ray structure of GM-CSF in complex with its re-
ceptor’s ectodomain, determined by Angel Lopez and
Section 19-3. Tyrosine Kinase–Based Signaling 717
Figure 19-46 Schematic model of Src activation. See the text
for an explanation.The coloring scheme largely matches that in
Fig. 19-45, as does the viewpoint. [After Young, M.A.,
P
P
P
P
P
P
P
X
X
P
P
P
P
Autoinhibited Form Active Form
SH3–SH2
linker
N
N
ATP
SH3
SH3
SH2
SH2
PTK domain
(N-lobe)
Lys 295
αC
αC
Glu 310
Glu 310
Tyr 416
Tyr 416
PTK domain
(C-lobe)
C
C
Tyr 527
Tyr 527
Src
activato
r
Substrate
Arg 409
Arg 409
Lys 295
Gonfloni, F., Superti-Furga, G., Roux, B., and Kuriyan, J., Cell
105, 115 (2001).]
JWCL281_c19_671-743.qxd 3/16/10 7:17 PM Page 717
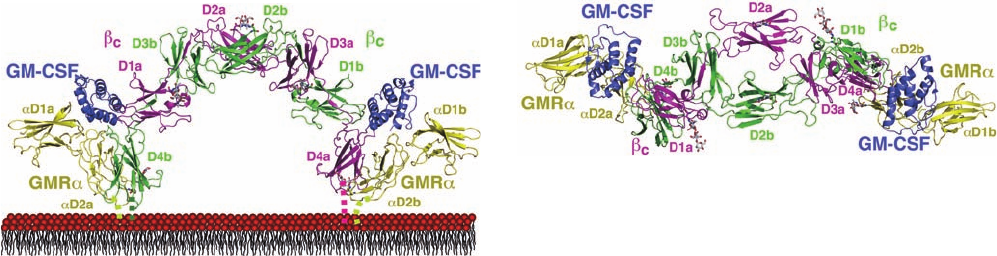
Michael Parker, reveals that it consists of a 2:2:2 complex
of GM-CSF, GMR, and
c
(Fig. 19-47). GM-CSF, as had
previously been determined, consists of an up–up–down–
down four-helix bundle, a topology that occurs only in
helical cytokines. Each GM-CSF molecule interacts with
both domains of a GMR subunit and with the N-terminal
domain of one
c
subunit (D1) and with the C-terminal do-
main of the other
c
subunit (D4).The latter phenomenon,
in which the domain from one subunit is exchanged with
the same domain from an identical subunit to form an in-
tertwined dimer, is known as domain swapping. It is a com-
mon mechanism for oligomer assembly.
Although the X-ray structures of the ectodomains of
several cytokine receptors have been determined, the
structures of their cytosolic domains are, as yet, unknown.
However, as James Darnell elucidated, the signal that heli-
cal cytokines have been bound by their cognate receptors
is transmitted within the cell by the JAK-STAT pathway.
These cytokine receptors form complexes with proteins of
the Janus kinase (JAK) family of NRTKs, so named be-
cause each of its four ⬃1150-residue members (JAK1,
JAK2, JAK3, and Tyk2) has two PTK domains (Janus is
the two-faced Roman god of gates and doorways),
although only the C-terminal domain is functional (Fig.
19-44). STATs (for signal transducers and activators of
transcription) comprise a family of seven ⬃800-residue
proteins that are the only known transcription factors
whose activities are regulated by Tyr phosphorylation and
that have SH2 domains.
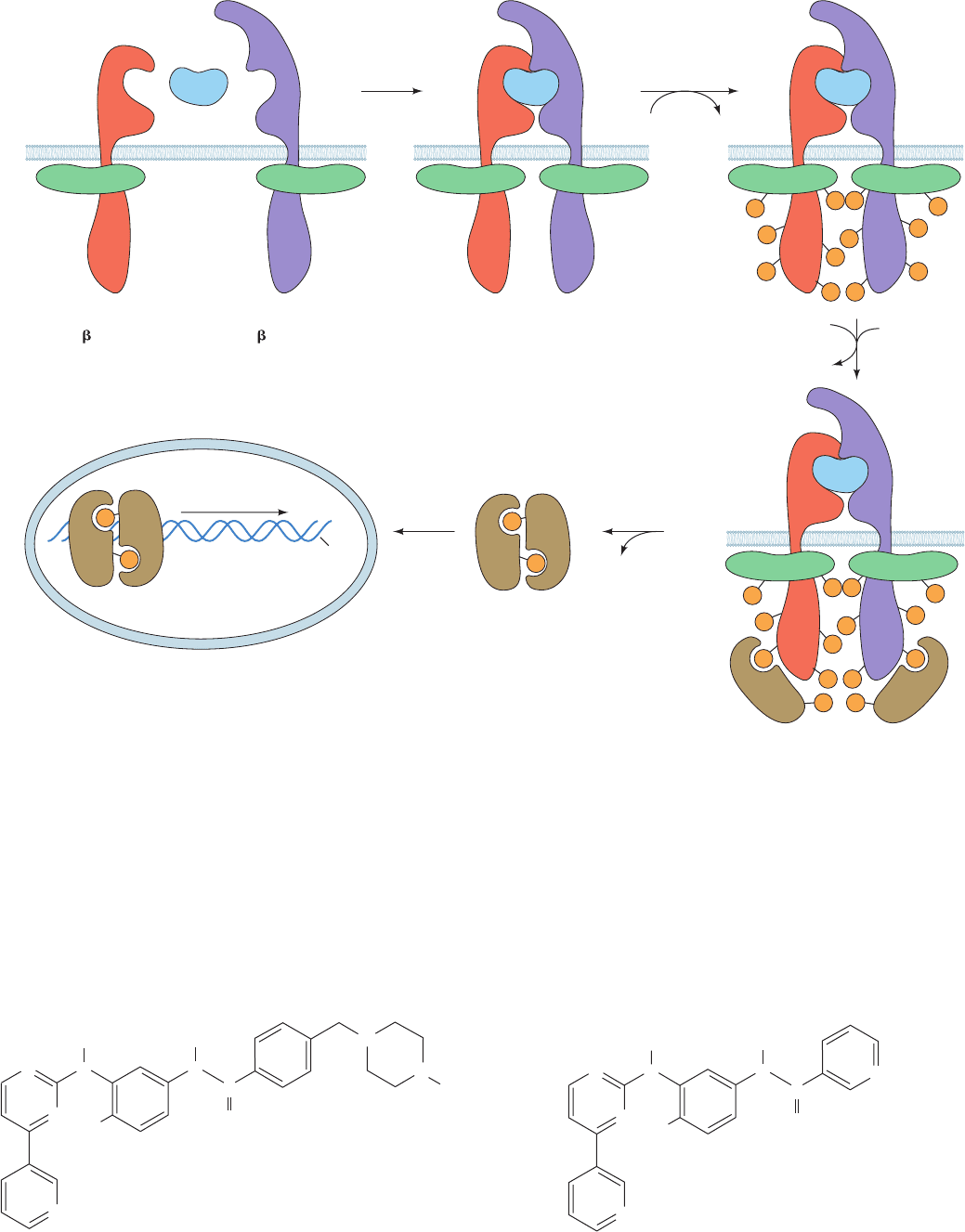
The JAK-STAT pathway functions as is diagrammed in
Fig. 19-48:
1. Cytokine binding induces the cytokine receptor to
oligomerize.
2. The cytokine receptor’s two associated JAKs are
thereby brought into apposition (in the case of the GM-CSF
receptor,JAK2 binds to the cytosolic domain of
c
), where-
upon they reciprocally phosphorylate each other and then
their associated receptors, a process resembling the au-
tophosphorylation of dimerized RTKs (Section 19-3Ab).
Note that unlike most NRTKs, JAKs lack both SH2 and
SH3 domains.
3. STATs bind to the phosphoTyr group on their cog-
nate activated receptor via their SH2 domain and are then
phosphorylated on a conserved Tyr residue by the associ-
ated JAK.
4. Following their dissociation from the receptor, the
phosphorylated STATs homo- or heterodimerize via the
association of their phosphoTyr residue with the SH2 do-
main on the opposing subunit.
5. The STAT dimers are translocated to the nucleus,
where these now functional transcription factors induce
the expression of their target genes in much the same way
as do the transcription factors that are phosphorylated by
the MAPKs (Fig. 19-40).
c. PTKs Are Targets of Anticancer Drugs
The hallmark of chronic myelogenous leukemia (CML)
is a specific chromosomal translocation (Section 34-4C)
forming the so-called Philadelphia chromosome in which
the Abl gene (which encodes the NRTK Abl) is fused with
the Bcr gene (which encodes the protein Ser/Thr kinase
Bcr). The Abl portion of this Bcr–Abl fusion protein is con-
stitutively (continuously, without regulation) activated,
probably because its Bcr portion oligomerizes. Hematopoi-
etic stem cells (from which all blood cells are descended)
bearing the Philadelphia chromosome are therefore primed
to develop CML (malignancy requires several independent
genetic alterations; Section 19-3Ba). Without a bone mar-
row transplant (a high-risk procedure that is unavailable to
718 Chapter 19. Signal Transduction
(a)
(b)
Figure 19-47 X-ray structure of the extracellular portions of
the GM-CSF receptor in complex with GM-CSF. The structure of
this 2-fold symmetric 2:2:2 complex of GM-CSF, GMR, and
c
is viewed (a) parallel to the plane of the plasma membrane and
(b) from the extracellular side of the membrane. The GM-CSF is
purple, the GMR is yellow, one
c
monomer (chain a) is
magenta and the other (chain b) is green.The labels indicate the
domain names.The observed N-linked carbohydrates are drawn
in stick form with Asn C green, carbohydrate C gray, N blue, and
O red.The disordered peptides that link the C-termini of the
receptor chains to their transmembrane segments are
represented by dashed lines. [Modified from a drawing by Angel
Lopez, Hanson Institute,Adelaide,Australia, and Michael
Parker, St. Vincent’s Institute of Medical Research, Fitzroy,
Victoria, Australia. PDBid 3CXE.]
JWCL281_c19_671-743.qxd 3/16/10 7:17 PM Page 718
which was developed by Brian Druker and Nicholas
Lydon, has caused the remission of symptoms in ⬃90% of
CML patients with almost no serious side effects. This un-
precedented performance occurs, in part, because Gleevec
is essentially inactive against other PTKs (an exception be-
ing the PDGF receptor) as well as protein Ser/Thr kinases.
Abl resembles Src (Fig. 19-44) but lacks Src’s C-terminal
regulatory phosphorylation site (Figs. 19-45 and 19-46).
The X-ray structure of Abl’s PTK domain in complex with
a truncated form of Gleevec,
N
N
N
C
O
H
3
C
H
N
N
N
H
Section 19-3. Tyrosine Kinase–Based Signaling 719
most individuals due to the lack of a suitable donor), CML
is invariably fatal, with an average survival time of ⬃6 years.
An inhibitor of Abl would be expected to prevent
the proliferation of, and even kill, CML cells. However,
to be an effective anti-CML agent, such a substance must
not inhibit other protein kinases because this would al-
most certainly cause serious side effects. Derivatives of
2-phenylaminopyrimidine bind to Abl with exceptionally
high affinity and specificity. One such derivative, imatinib
(trade name Gleevec),
N
N
N
N
C
O
CH
3
H
3
C
H
Imatinib (Gleevec
TM
)
N
N
N
H
Figure 19-48 The JAK-STAT pathway for the intracellular relaying of cytokine signals. See the
text for details. [After Carpenter, L.R., Yancopoulos, G.D., and Stahl, N., Adv. Protein Chem. 52,
109 (1999).]
P
P P
P
P
P
P
P
PP
PP
P P
P
P P
P
P
P
P
P
PP
P P
P
P
STAT
JAK JAK
STAT
STAT
STAT
P
P
STAT
STAT
1
45
ADPATP
2
ADP
ATP
3
JAK JAKJAK JAK
1
β
βββ β
β
β
β
122 21
21
JAK JAK
DNA
Gene expression
Nucleus
Extracellular space
Plasma
membrane
Cytoplasm
Cytokine receptor
1
subunit
Cytokine receptor
2
subunit
Cytokine
+
+
Cytokine
+
JAK
+
Receptors
STAT
JWCL281_c19_671-743.qxd 6/11/10 6:57 AM Page 719
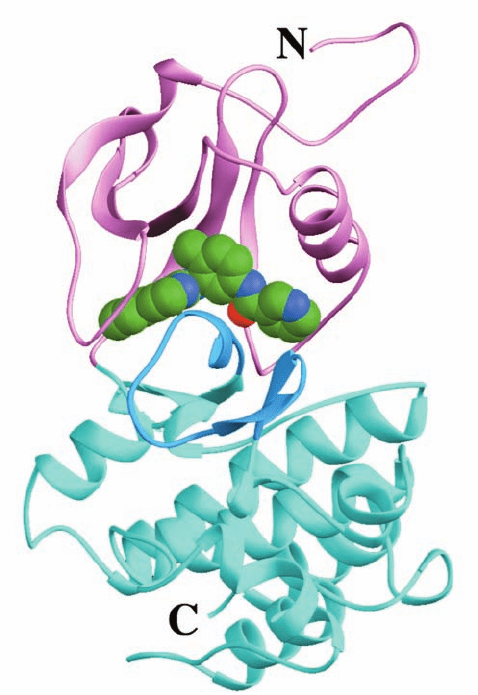
determined by Kuriyan (Fig. 19-49), reveals, as expected,
that the truncated Gleevec binds in Abl’s ATP-binding site
[the piperazinyl group that this inhibitor lacks relative to
Gleevec does not significantly alter its target discrimina-
tion but does increase Gleevec’s solubility and hence its
bioavailability (Section 15-4Ba); it probably binds in a sol-
vent accessible groove at the back of Abl]. Abl thereby
adopts an inactive conformation in which its activation
loop, which is not phosphorylated, appears to mimic the
way in which substrate peptides bind to PTKs (such as the
insulin receptor, Fig. 19-28a); that is, the activation loop as-
sumes an autoinhibitory conformation. As a consequence,
the N-terminal end of the activation loop, which has the
highly conserved sequence Asp-Phe-Gly (whose Asp side
chain, in the active PTK, ligates an Mg
2
ion that is essen-
tial for catalysis), assumes a conformation that is quite
different from that observed in the X-ray structures of
inactive Abl and Src (Fig. 19-45) because this latter confor-
mation would block the binding of Gleevec.
Gleevec was the first of several compounds that inhibit
specific protein kinases to be approved by the FDA for
clinical use against certain cancers. In addition, several
monoclonal antibodies (Section 6-1Da) that bind to spe-
cific PTKs or their ligands are in clinical use as anticancer
agents. For example, cetuximab (trade name Erbitux), a
chimeric mouse/human monoclonal antibody, is effective
against colorectal and certain head and neck cancers. It
specifically binds to the ectodomain of EGFR so as to
block its ligand binding and hence prevent its activation,
thus resulting in impaired cell growth and proliferation.
Such receptor-targeted therapies hold enormous promise
for controlling, if not curing, cancers by specifically target-
ing the aberrant proteins that cause the cancers. In con-
trast, most chemotherapeutic agents that are presently in
use indiscriminately kill fast-growing cells and hence al-
most always have debilitating side effects.
d. Increased Chaperone Activity Facilitates Cancer
Epidemiological studies have shown that individuals with
age-related neurodegenerative diseases such as Alzheimer’s
and Parkinson’s diseases have a far lower incidence of can-
cer than the general population. What is the biochemical
basis of this intriguing observation? These neurodegena-
tive diseases are all characterized by the deposition of
plaques containing amyloid fibrils (Section 9-5) in the af-
fected brain cells. In Alzheimer’s disease, these fibrils con-
sist mainly of A protein (Section 9-5B) and in Parkinson’s
disease they contain mainly -synuclein (a 140-residue sol-
uble protein of unknown function that normally occurs in
presynaptic terminals). These amyloid fibrils apparently
form because of age-associated reductions in the levels of
activity of the chaperone proteins that normally prevent
their aggregation.
Many oncogene products, as we have seen, are mutant
forms of proteins that participate in signal transduction and
hence have altered functionality. Such mutant proteins are
usually less stable than their unmutated forms and hence re-
quire greater than normal attention from chaperone pro-
teins to maintain their active conformations. In particular,
the Hsp90 chaperone proteins (Section 9-2C) facilitate the
late stage folding of numerous signaling proteins and, pre-
sumably, their oncogenic mutants. In fact, many unmutated
signaling proteins are unstable unless they are binding their
corresponding ligand or protein and hence are present in
cells in complex with Hsp90 (which accounts for Hsp90’s
high abundance, normally 1–2% of a cell’s soluble proteins).
Hsp90 is overexpressed in many types of cancers, a situ-
ation that is correlated with resistance to therapy and
hence a poor prognosis (and which accounts for the obser-
vation that individuals with neurodegenerative diseases
rarely get cancer). Consequently, Hsp90 inhibitors are anti-
cancer drugs. Several Hsp90 inhibitors, all of which inter-
fere with its ATPase function, are presently in clinical trials.
These substances, unlike most targeted anticancer drugs
(e.g., Gleevec), are likely to be effective against a broad
range of cancers.
720 Chapter 19. Signal Transduction
Figure 19-49 X-ray structure of the Abl PTK domain in
complex with a truncated derivative of Gleevec. The protein is
viewed from the right of the “standard” view of protein kinases
(e.g., Figs. 19-28a and 19-45), with its N-terminal lobe pink, its
C-terminal lobe cyan, and its activation loop blue. The truncated
Gleevec, which occupies the PTK’s ATP-binding site, is shown in
space-filling form with C green, N blue, and O red. [Based on an
X-ray structure by John Kuriyan, The Rockefeller University.
PDBid 1FPU.]
JWCL281_c19_671-743.qxd 3/16/10 7:17 PM Page 720