Voet D., Voet Ju.G. Biochemistry
Подождите немного. Документ загружается.

Section 19-3. Tyrosine Kinase–Based Signaling 701
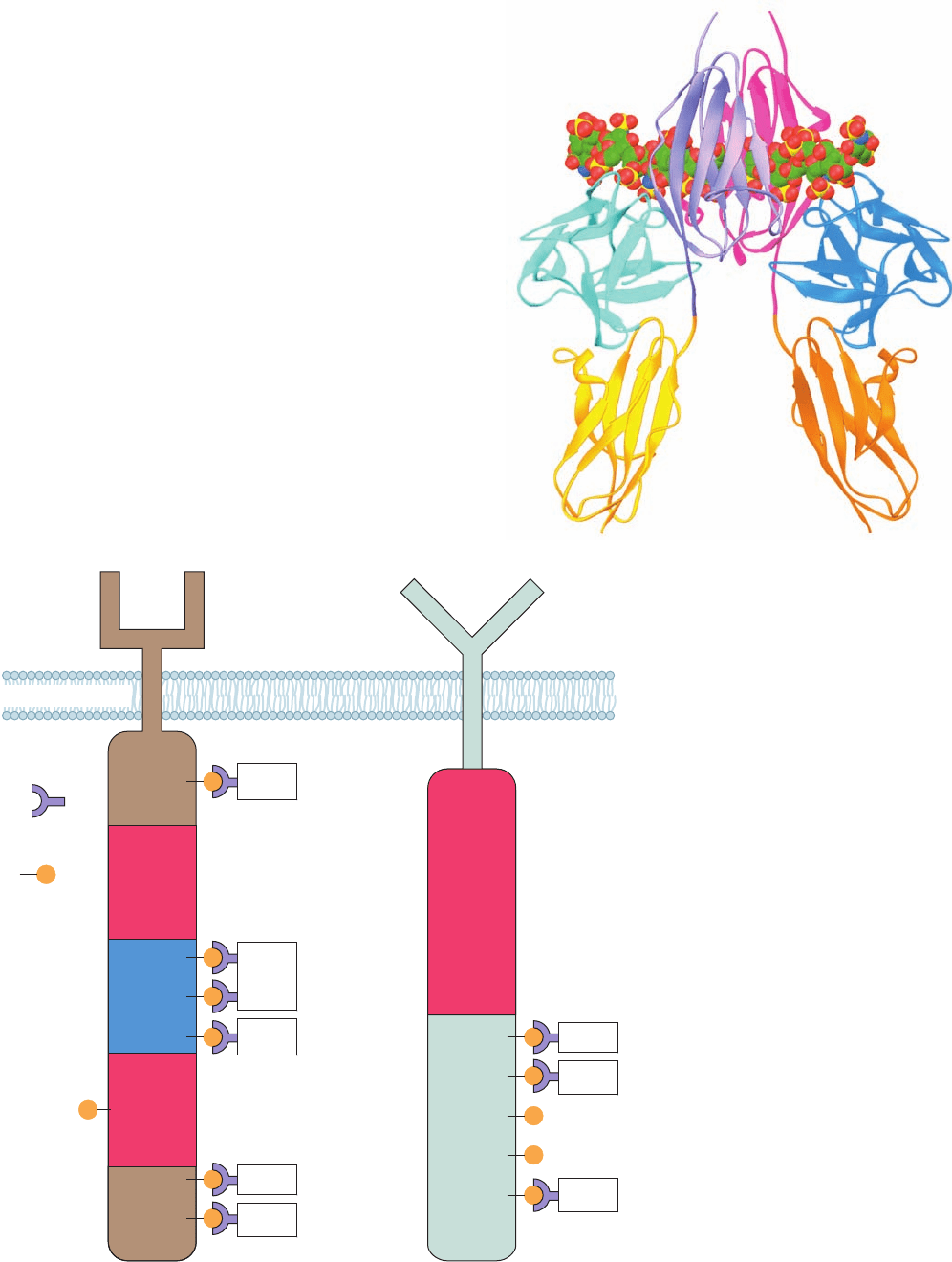
Figure 19-27 Schematic diagrams of
RTKs. (a) The PDGF receptor and (b) the
EGF receptor.Their autophosphorylation
sites and the proteins that are activated by
binding to these sites via their SH2 domains
(all of which are discussed in this chapter)
are indicated. Note that almost all of the
autophosphoryated Tyr residues that bind
to other proteins lie outside the tyrosine
kinase domains. [After Pawson, T. and
Schlessinger, J., Curr. Biol. 3, 435 (1993).]
Plasma membrane
Extracellular
Cytosol
SH2 domain
Tyrosine
kinase
domain
Tyrosine
kinase
domain
Tyrosine
kinase
domain
Kinase
insert
sequence
Phospho-Tyr
residue
PY
Y
PY
PY
PY
PY
PY
PY
PI3
kinase
RasGAP
P Src
PDGF
receptor
EGF
receptor
SHP-2
PLC-γ 1
PY
PY
PY
PY
PY
PLC-γ 1
Grb2
Shc
(a) (b)
Figure 19-26 X-ray structure of the 2:2:2 complex of FGF2, the D2–D3 portion of FGFR1,
and a heparin decasaccharide. The view is normal to the dimeric complex’s 2-fold axis of
symmetry, with the plasma membrane below. The polypeptides are represented in
ribbon form with the FGF molecules cyan and blue, the D2 and D3
domains of one FGFR monomer lavender and gold, and those of the other
magenta and orange. The two heparin decasaccharides are shown in
space-filling form with C green, N blue, O red, and S yellow. The D2
and D3 domains of FGFR each have an immunoglobulin fold: a 
sandwich comprised of a 3-stranded and a 4-stranded antiparallel
 sheet (Section 8-3Bg). [Based on an X-ray structure by Moosa
Mohammadi, New York University School of Medicine. PDBid 1FQ9.]
JWCL281_c19_671-743.qxd 6/4/10 10:53 AM Page 701
19-3D, this causes the activation of the proteins that partic-
ipate in executing the instructions implied by the extracel-
lular presence of the protein growth factor.
c. The Insulin Receptor PTK Undergoes Major
Conformational Changes on Autophosphorylation
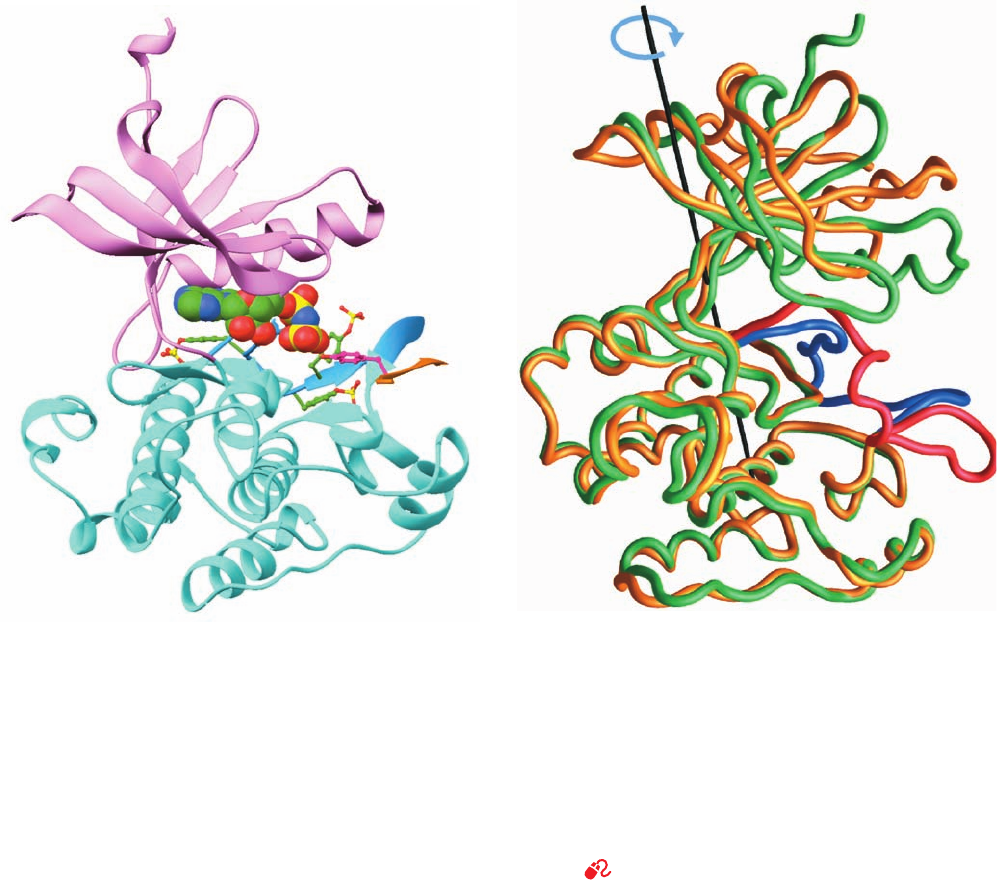
How does autophosphorylation activate a PTK? The
comparison of the X-ray structures of the PTK domain of
the insulin receptor in its inactive unphosphorylated and
active triphosphorylated states has done much to answer
this question. The insulin receptor is expressed as a single
1382-residue precursor peptide that is proteolytically
processed to yield the disulfide-linked ␣ and  subunits
(731 and 619 residues) of the mature receptor (Fig. 19-25).
The X-ray structure of a 306-residue PTK-containing seg-
ment of the  subunit that was phosphorylated at its three
autophosphorylation sites, Tyr residues 1158, 1162, and
1163 (using the precursor numbering system), and in com-
plex with the nonhydrolyzable ATP analog AMPPNP and
an 18-residue peptide substrate, was determined by Stevan
Hubbard. It reveals (Fig. 19-28a) that this PTK structurally
resembles other PTKs of known structure as well as pro-
tein Ser/Thr kinases such as PKA (Fig. 18-15) and the phos-
phorylase kinase ␥ subunit (Fig. 18-21).
Comparison of this structure with that of the unphospho-
rylated and uncomplexed protein, determined by Hubbard
and Wayne Hendrickson, reveals that, on phosphorylation
and substrate binding,the PTK’s N-terminal lobe undergoes a
nearly rigid 21° rotation relative to the C-terminal lobe
about the long axis of the protein (Fig. 19-28b). This dra-
matic conformational change closes the active site cleft
about the AMPPNP and, presumably, correctly positions
702 Chapter 19. Signal Transduction
(a)
(b)
Figure 19-28 X-ray structure of the PTK domain of the
insulin receptor. (a) The PTK phosphorylated at Tyr residues
1158, 1162, and 1163 and in complex with AMPPNP and an
18-residue polypeptide substrate. The PTK is shown in the
“standard” protein kinase orientation with its N-terminal domain
pink, its C-terminal domain cyan, and its activation loop light
blue. Its three phosphorylated Tyr side chains are shown in
ball-and-stick form with C green, N blue, O red, and P yellow.
The AMPPNP, which is identically colored, is shown in
space-filling form.The substrate polypeptide is orange (only 6 of
its residues are visible) and its phosphorylatable Tyr residue is
shown in ball-and-stick form with C magenta and O red. (b) The
polypeptide backbones of the phosphorylated and
unphosphorylated forms of the insulin receptor PTK domain as
superimposed on their C-terminal lobes.The phosphorylated
protein is green with its activation loop blue, and the
unphosphorylated protein is yellow with its activation loop red.
The axis (black line) and arrow (blue) indicate the rotation
required to align the N-terminal domain of the unphosphorylated
protein with that of the phosphorylated protein. [Part a based on
an X-ray structure by and Part b courtesy of Stevan Hubbard,
New York University Medical School. PDBids 1IR3 for the
phosphorylated protein and 1IRK for the unphosphorylated
protein.]
See Interactive Exercise 13
JWCL281_c19_671-743.qxd 10/19/10 7:36 AM Page 702
critical residues for substrate binding and catalysis. All
three phosphorylated Tyr residues occur on the PTK’s acti-
vation loop (residues 1149–1170; recall that many Ser/Thr
kinases are also phosphorylated on their activation loops;
Section 18-3Cb). The unphosphorylated activation loop
threads through the PTK’s active site so as to prevent the
binding of both ATP and protein substrates. However, on
phosphorylation, the activation loop assumes a conforma-
tion that does not occlude the active site (Fig. 19-28b) but
instead forms part of the substrate recognition site. The
phosphate group on Tyr 1163 bridges the activation loop by
forming a hydrogen bond with the side chain of the con-
served Arg 1155 on one side of the loop and with the main
chain N of Gly 1166 on its other side.The phosphate group
of Tyr 1162 makes two hydrogen bonds to the side chain of
the conserved Arg 1164. However, the phosphate group of
Tyr 1168 makes no protein contacts, which suggests that it
forms a docking site for downstream signaling proteins
(see below). These observations are in agreement with ex-
periments indicating that the tyrosine kinase activity of the
insulin receptor increases with the degree of phosphoryla-
tion at its three autophosphorylatable Tyr side chains and
that full activity is not achieved until Tyr 1163 is phospho-
rylated. Indeed, nearly all known RTKs have between one
and three autophosphorylatable Tyr residues in their acti-
vation loops (a major exception being the members of
EGFR subfamily; Fig. 19-27b) and, in all phosphorylated
protein kinases of known structure, assume similar confor-
mations.
Only the 6 centrally located residues, GDYMNM, of
the 18-residue substrate peptide are seen in the foregoing
X-ray structure. These include the YMXM sequence found
in all efficient substrates of the insulin receptor. This seg-
ment associates with the kinase as a strand in a sheet. Its
Met side chains fit into adjacent hydrophobic pockets of
the protein, whereas its phosphorylatable Tyr side chain ex-
tends toward the phosphate group of the AMPPNP (Fig.
19-28a). The specificity of the protein for phosphorylating
Tyr rather than Ser or Thr is explained by the observation
that the side chain of Tyr, but not those of Ser or Thr, are
long enough to reach the active site.
B. Cancer: The Loss of Control of Growth
Before we continue our discussion of signaling pathways,
let us consider cancer, a group of diseases that is character-
ized by defects in signal transduction causing uncontrolled
growth. Indeed, studies of cancer have greatly increased
our understanding of signal transduction and vice versa.
The cells of the body normally remain under strict devel-
opmental control. For instance, during embryogenesis, cells
must differentiate, proliferate, migrate, and even die in the
correct spatial arrangement and temporal sequence to
yield a normally functioning organism. In the adult, the
cells of certain tissues, such as the intestinal epithelium, the
blood-forming tissues of the bone marrow, and those of
hair follicles, continue to proliferate. Most adult body cells,
however, have permanently ceased doing so.
Cells occasionally lose their developmental controls and
commence excessive proliferation. The resulting tumors
can be of two types:
1. Benign tumors, such as warts and moles, grow by sim-
ple expansion and often remain encapsulated by a layer of
connective tissue. Benign tumors are rarely life threaten-
ing, although if they occur in an enclosed space such as in
the brain or secrete large quantities of certain hormones,
they can be lethal.
2. Malignant tumors or cancers grow in an invasive
manner and shed cells that, in a process known as metasta-
sis, colonize new sites in the body. Malignant tumors are al-
most invariably life threatening; they are responsible for
20% of the mortalities in the United States.
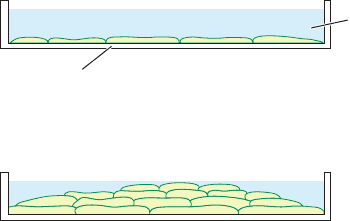
The most obvious and the medically most significant
property of cancer cells is that they proliferate uncontrol-
lably. For instance, when grown in a tissue culture dish, nor-
mal cells form a monocellular layer on the bottom of the
dish and then, through a process termed contact inhibition,
cease dividing (Fig. 19-29a). In contrast, the growth of ma-
lignant cells is unhampered by intercellular contacts; in cul-
ture they form multicellular layers (Fig. 19-29b). Moreover,
even in the absence of contact inhibition, normal cells are
far more limited in their capacity to reproduce than are
cancer cells. Normal cells, depending on the species and age
of the animal from which they were taken, will only divide
in culture 20 to 60 times before they reach senescence (a
stage at which they cease dividing) and die (a phenomenon
that, no doubt, is at the heart of the aging process; Section
30-4Db). Cancer cells, on the other hand, are immortal; there
is no limit to the number of times they can divide. In fact,
some cancer cell lines have been maintained in culture
through thousands of divisions spanning 6 decades. Immor-
tal cells, however, are not necessarily malignant: The hall-
mark of cancer is immortality combined with uncontrolled
growth.
Section 19-3. Tyrosine Kinase–Based Signaling 703
Figure 19-29 Growth pattern of vertebrate cells in culture.
(a) Normal cells stop growing through contact inhibition once they
have formed a confluent monolayer. (b) In contrast, transformed
cells lack contact inhibition; they pile up to form a multilayer.
Plastic
tissue culture
dish
(a) Normal cells
(b) Transformed cells
Growth medium
JWCL281_c19_671-743.qxd 3/16/10 7:16 PM Page 703
a. Cancer Is Caused by Carcinogens,
Radiation, and Viruses
Most cancers are caused by agents that damage DNA or
interfere with its replication or repair. These include a
great variety of man-made and naturally occurring sub-
stances known as chemical carcinogens (Section 30-5F),
as well as radiation, both electromagnetic and particulate,
with sufficient energy to break chemical bonds. In addi-
tion, certain viruses induce the formation of malignant
tumors in their hosts (see below).
Almost all malignant tumors result from the transfor-
mation of a single cell [conversion to the cancerous state;
this term should not be confused with the acquisition of
genetic information from exogenously supplied DNA
(Section 5-2A)], which, then being free of its normal de-
velopmental constraints, proliferates. Yet, considering, for
example, that the human body consists of around 10
14
cells,
transformation must be a very rare event. One of the major
reasons for this, as the age distribution of the cancer death
rate indicates (Fig. 19-30), is that transformation requires a
cell or its ancestors to have undergone several independent
and presumably improbable carcinogenic changes. Conse-
quently, exposure to a carcinogen may prime many cells for
transformation, but a malignant tumor may not form until
decades later when one of these cells suffers a final trans-
forming event.
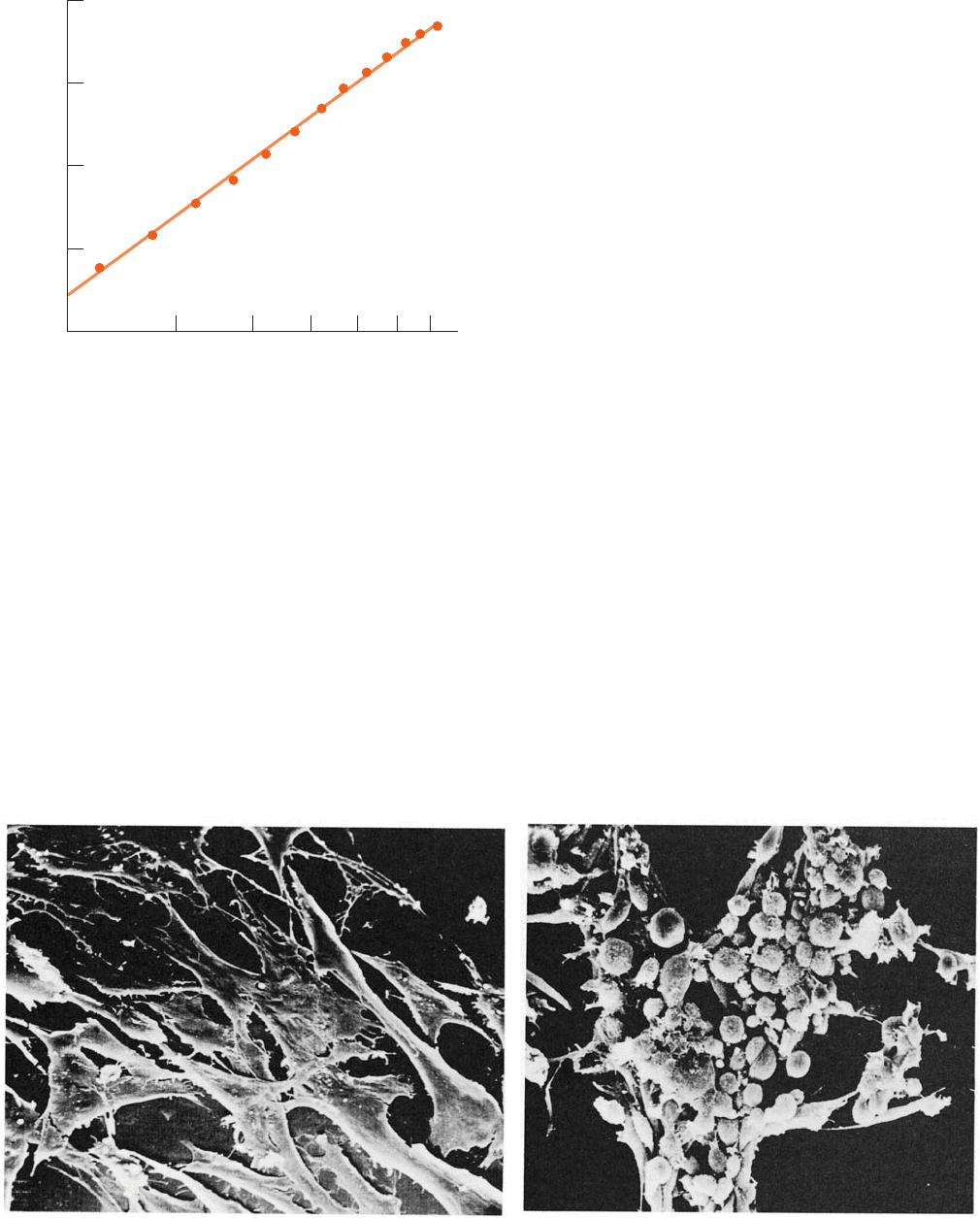
The viral induction of cancer was first observed in 1911
by Peyton Rous, who demonstrated that cell-free filtrates
from certain chicken sarcomas (malignant tumors arising
from connective tissues) promote new sarcomas in chick-
ens (Fig. 19-31). Although decades were to pass before the
significance of this work was appreciated (Rous was
awarded the Nobel prize in 1966 at the age of 85), many
other such tumor viruses have since been characterized.
The Rous sarcoma virus (RSV), as are all known RNA tu-
mor viruses, is a retrovirus (an RNA virus that replicates its
chromosome by copying it to DNA in a process mediated
by a virally encoded reverse transcriptase, inserting the
DNA into the host’s genome, and then transcribing this
DNA). It contains a gene, v-src (“v” for viral, “src” for sar-
coma), which encodes a protein named v-Src that mediates
704 Chapter 19. Signal Transduction
Figure 19-30 Variation of the cancer death rate in humans
with age. The linearity of this log–log plot can be explained by
the hypothesis that several randomly occurring mutations are
required to generate a malignancy. The slope of the line suggests
that, on the average, five such mutations are required for a
malignant transformation.
Figure 19-31 Transformation of cultured chicken fibroblasts
by Rous sarcoma virus. (a) Normal cells adhere to the surface of
the culture dish, where they assume a flat extended
20 8030 50 706040
Annual death rate/milliion
Age
1
10
100
1000
(a)
(b)
conformation. (b) On infection with RVS, these cells become
rounded and cluster together in piles. [Courtesy of G. Steven
Martin, University of California at Berkeley.]
JWCL281_c19_671-743.qxd 3/16/10 7:16 PM Page 704
host cell transformation. v-src has therefore been termed
an oncogene (Greek: onkos, mass or tumor).
What is the origin of v-src and what is its viral function?
Hybridization studies (Section 5-3Cb) by Michael Bishop
and Harold Varmus in 1976 led to the remarkable discov-
ery that uninfected chicken cells contain a gene, c-src (“c”
for cellular), that is homologous to v-src. Moreover, c-src is
highly conserved in a wide variety of eukaryotes that span
the evolutionary scale from Drosophila to humans.This ob-
servation strongly suggests that c-src, which antibodies di-
rected against v-Src indicated is expressed in normal cells,
is an essential cellular gene. In fact, both v-Src and its
normal cellular analog, c-Src, function to stimulate cell
proliferation (Section 19-3Ea). Apparently, v-src was origi-
nally acquired from a cellular source by an initially non-
transforming ancestor of RSV. By maintaining the host
cell in a proliferative state (cells are usually not killed by
RSV infection), v-Src presumably enhances the viral repli-
cation rate.
b. Viral Oncogene Products Mimic the Effects of
Protein Growth Factors and Hormones
The proteins encoded by many viral oncogenes are
analogs of various growth factor and hormone system com-
ponents. For instance:
1. The v-sis oncogene of simian sarcoma virus encodes
a protein secreted by infected cells that is nearly identical
to PDGF. Hence, the uncontrolled growth of simian sar-
coma virus–infected cells apparently results from the
continuous and inappropriate presence of this PDGF
homolog.
2. Nearly half of the more than 20 known retroviral
oncogenes, including v-src, encode PTKs. For example, the
v-erbB oncogene specifies a truncated version of the EGF
receptor (Fig. 19-27b) that lacks the EGF-binding domain
but retains its transmembrane segment and its protein ki-
nase domain. Evidently, oncogene-encoded PTKs inappro-
priately phosphorylate the target proteins normally recog-
nized by RTKs, thereby driving the afflicted cells to a state of
unrestrained proliferation.
3. The v-ras oncogene encodes a protein, v-Ras, that
functionally resembles the monomeric G-protein c-Ras
(Section 19-3Cf) in that it is localized on the cytoplasmic
side of the mammalian plasma membrane where, when
binding GTP, it activates a variety of cellular processes by
stimulating the phosphorylation of numerous proteins at
specific Ser and Thr residues. Although v-Ras hydrolyzes
GTP to GDP, it does so much more slowly than c-Ras. The
restraint to protein phosphorylation that GTP hydrolysis
would normally impose on c-Ras is thus greatly reduced in
v-Ras, thereby transforming the cell.
4. Several viral oncogenes, including v-jun and v-fos,
encode nuclear proteins whose corresponding normal cellu-
lar analogs are synthesized in response to growth factors
such as EGF and PDGF that induce mitosis (cell division).
Many such proteins, including the v-jun and v-fos gene prod-
ucts, bind to DNA, strongly suggesting that they influence
its transcription and/or replication. Indeed, v-jun is 80%
identical in sequence to the proto-oncogene (normal cellu-
lar analog of an oncogene) c-jun, which encodes a tran-
scription factor named Jun (also called AP-1; Section 19-
3D). Moreover, Jun/AP-1 forms a tight complex with the
protein encoded by the proto-oncogene c-fos, which
greatly increases the ability of Jun/AP-1 to stimulate tran-
scription from Jun-responsive genes.
Oncogene products therefore appear to be functionally
modified or inappropriately expressed components of elab-
orate control networks that regulate cell growth and differ-
entiation. The complexity of these networks (as we shall
see, cells generally respond to a variety of growth factors,
hormones, and transcription factors in partially overlap-
ping ways) is probably why malignant transformation re-
quires several independent carcinogenic events. Note,how-
ever, that few human cancers are virally induced; nearly all
of them arise from genetic alterations involving proto-
oncogenes. We discuss the nature of these alterations in
Section 34-4C.
C. Relaying the Signal: Binding Modules, Adaptors,
GEFs, and GAPs
Many autophosphorylated RTKs can directly phosphory-
late their target proteins. Surprisingly, however, not all
RTKs do so. How, then, do they activate their target pro-
teins? The answer, as we shall see, is through a highly di-
verse and complicated set of interconnected signaling
pathways involving cascades of associating proteins.
a. Two-Hybrid Systems Identify Proteins That
Interact in vivo
Before we consider the interacting proteins that partici-
pate in RTK-mediated signal transduction, let us discuss
one of the most often used methods to detect their associa-
tions in vivo, the two-hybrid system. This ingenious experi-
mental technique, which was formulated by Stanley Fields,
is based on the peculiar bipartite nature of many transcrip-
tion factors (proteins that bind to the promoters and other
upstream control regions of eukaryotic genes and, in doing
so, influence the rate at which RNA polymerase initiates
the transcription of these genes; Section 5-4Ab). Such tran-
scription factors, as we further discuss in Section 34-3Bi,
contain a DNA-binding domain (DBD) that targets the
transcription factor to a specific DNA sequence and an ac-
tivation domain (AD) that recruits RNA polymerase to
initiate transcription at a nearby transcriptional initiation
site. These two domains function independently, so that a
genetically engineered hybrid protein with the DBD of
one transcription factor and the AD of another will acti-
vate the transcription of the gene for which the DBD is tar-
geted. Moreover, it makes little difference as to whether
the DBD is on the N-terminal or the C-terminal side of the
AD, regardless of how they are arranged in their parent
proteins. Evidently, as long as a DBD and an AD are held
in proximity, they function as a transcription factor for the
gene(s) to which the DBD is targeted.
Section 19-3. Tyrosine Kinase–Based Signaling 705
JWCL281_c19_671-743.qxd 3/16/10 7:16 PM Page 705
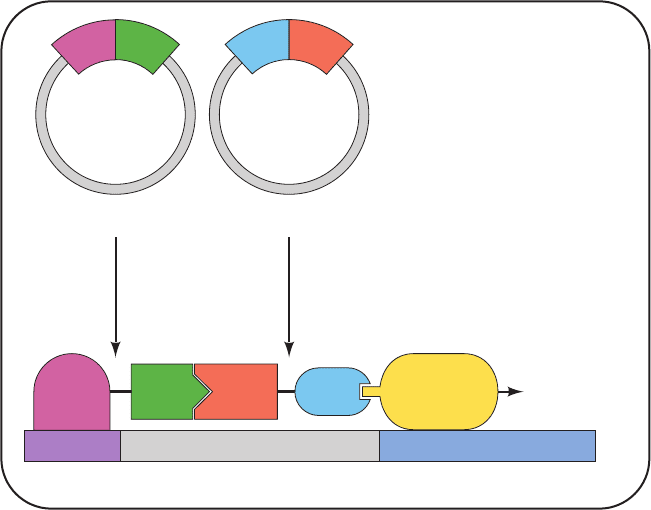
The two-hybrid system employs two different plasmids
in yeast (Fig. 19-32): One encodes a hybrid protein consist-
ing of a DBD fused to a so-called bait or probe protein; the
other encodes a hybrid protein consisting of an AD fused
to a so-called fish or target protein.The DBD is targeted to
a reporter gene that has been engineered into the yeast
chromosome such as the E. coli lacZ gene, which encodes
the enzyme -galactosidase. On culture plates containing
X-gal (a colorless compound that turns blue on being hy-
drolyzed by -galactosidase; Section 5-5Ca), yeast colonies
expressing -galactosidase turn blue, thereby indicating
that the bait and fish proteins they encode associate with
one another. Using this technique,cells can be screened for
fish proteins that specifically interact with a particular bait
protein by properly inserting the various cDNAs (Section
5-5F) derived from the cells into the AD-containing plas-
mid. A fish protein selected in this way can then be identi-
fied by sequencing its cDNA.
b. SH2 Domains Mediate Signal Transduction
Now let us return to our discussion of RTK-mediated
signaling. Many (100) of the diverse cytoplasmic proteins
that bind to autophosphorylated receptors, for example,
the cytoplasmic PTK c-Src (henceforth referred as just
Src), certain GTPase activating proteins (GAPs), and
phospholipase C- (Section 19-4Bc), contain one or two
conserved ⬃100-residue modules known as Src homology
domain 2 (SH2; so named because they were first noticed
in tyrosine kinases related to Src; SH1 refers to their cat-
alytic domains). SH2 domains specifically bind phosphoTyr
residues in their target peptides with high affinity; they bind
their unphosphorylated target peptides weakly if at all.
Most of the phosphoTyr residues to which SH2 binds are
located in the juxtamembrane (just after the transmem-
brane helix), kinase insert, and C-terminal regions of
RTKs; those in the activation loop function mainly to stim-
ulate PTK activity. Indeed, RTK autophosphorylation oc-
curs in two phases: First the activation loops of the RTK
are phosphorylated and then the resulting activated PTK
phosphorylates the other sites on the opposing subunit of
the RTK.
The X-ray and NMR structures of SH2 domains from
several proteins, both alone and in complex with phospho-
Tyr (pY)-containing polypeptides, have been determined.
SH2 is a hemispherically shaped domain, which contains a
central 5-stranded antiparallel sheet that is sandwiched
between two nearly parallel helices.The N- and C-terminal
residues of SH2 are in close proximity on the surface oppo-
site the peptide-binding site, which suggests that this do-
main can be inserted between any two surface residues on
a protein without greatly disturbing its fold or function. In-
deed, the sequences of a variety of SH2-containing pro-
teins reveal no apparent preference for the location of this
domain in a protein.
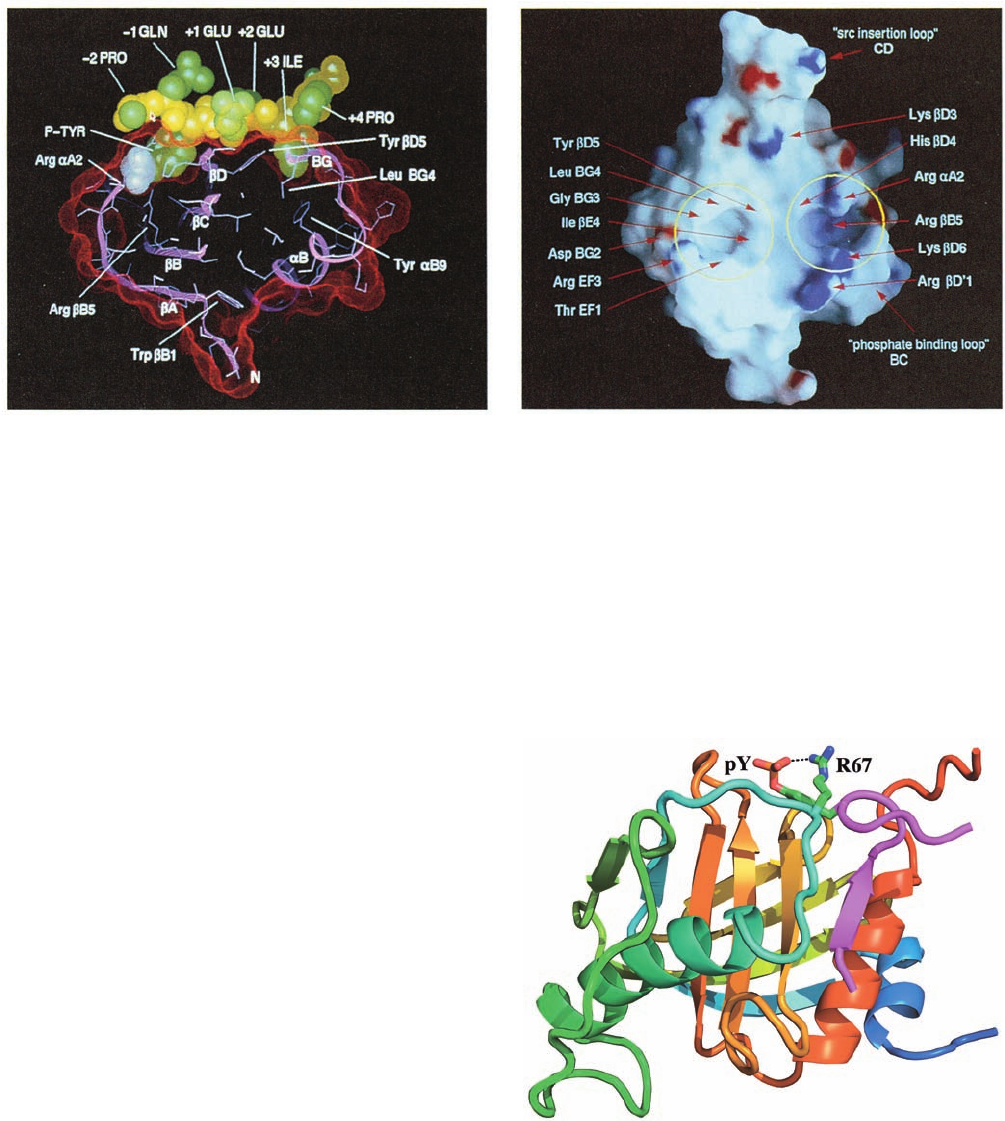
John Kuriyan determined the X-ray structure of the
SH2 domain of Src in complex with an 11-residue polypep-
tide containing the sequence pYEEI, a tetrapeptide seg-
ment that binds to this SH2 domain with high affinity. The
11-residue peptide binds to the SH2 domain in an extended
conformation, with contact being made primarily by the
pYEEI tetrapeptide (Fig. 19-33a). The phosphoTyr side
chain inserts into a small cleft formed, in part, by three
highly conserved positively charged residues, including the
side chain of an invariant Arg, which contacts the phos-
phate group. The Ile side chain is similarly inserted into a
nearby hydrophobic pocket and the entire tetrapeptide
segment interacts very tightly with SH2, although the side
chains of the peptide’s two central Glu residues do not
706 Chapter 19. Signal Transduction
Figure 19-32 The two-hybrid system.
Two plasmids are expressed in yeast: One
encodes a hybrid protein consisting of the
DNA-binding domain (DBD) of a
transcription factor fused to a “bait”
protein, and the other encodes the
activation domain (AD) of a transcription
factor fused to a “fish” protein. The DBD
specifically binds to the promoter of a
reporter gene, here lacZ. If the “fish”
protein associates with the “bait” protein,
its attached AD recruits RNA polymerase
to initiate the transcription of the reporter
gene. The -galactosidase encoded by the
lacZ gene is readily detected through the
use of X-gal, which turns blue when
hydrolyzed by -galactosidase. Yeast
colonies expressing “bait” and “fish”
proteins that do not interact remain
colorless, as do colonies that do not express
both plasmids.
Yeast
cell
Plasmid 1 Plasmid 2
Expression
Promoter
DBD Bait Fish
lacZ reporter gene
Transcriptional
initiation
RNA
polymerase
D
B
D
B
a
i
t
AD
A
D
F
i
s
h
JWCL281_c19_671-743.qxd 3/16/10 7:16 PM Page 706
project toward SH2. Thus, the peptide resembles a two-
pronged plug that is inserted into a two-holed socket on
SH2 (Fig.19-33b). Comparison of this structure with that of
uncomplexed Src SH2 indicates that, on binding peptide,
SH2 undergoes only small conformational changes that are
localized at its peptide-binding site. These structures pro-
vide a simple explanation for why SH2 does not bind the
far more abundant phosphoSer- and phosphoThr-containing
peptides: The side chains of these residues are too short
for their phosphate groups to contact the invariant Arg
side chain at the bottom of the phosphoTyr-binding pocket.
c. PTB Domains Also Bind PhosphoTyr-
Containing Peptides
A second type of motif that specifically binds to
phosphoTyr-containing target peptides is known as the
phosphotyrosine-binding (PTB) domain. PTB domains
specifically bind the consensus sequence NPXpY (where X
is any residue) and hence recognize the sequence on the
N-terminal side of pY rather than that on its C-terminal
side, as do SH2 domains.
The NMR structure of the 195-residue PTB domain of
Shc, an adaptor protein (see below), in complex with a
12-residue target peptide containing the centrally located
sequence NPQpY was determined by Stephen Fesik. The
structure consists of a  sandwich comprising two nearly
perpendicular antiparallel  sheets flanked by three ␣ he-
lices (Fig. 19-34). The N-terminal segment of the target
phosphopeptide assumes an extended conformation that,
Section 19-3. Tyrosine Kinase–Based Signaling 707
(a)
(b)
Figure 19-33 X-ray structure of the 104-residue Src SH2
domain in complex with an 11-residue polypeptide
(EPQpYEEIPIYL) containing the protein’s pYEEI target
tetrapeptide. (a) A cutaway view of the complex, in which its
solvent-accessible surface is represented by red dots, the protein
(pink) is shown in ribbon form with its side chains in stick form
(light blue), and the N-terminal 8-residue segment of the bound
polypeptide is shown in space-filling form with its backbone
yellow, its side chains green, and its phosphate group white [the
Figure 19-34 The NMR structure of the PTB domain of Shc
in complex with a 12-residue polypeptide (HIIENPQpYFSDA)
from the Shc binding site of a nerve growth factor (NGF)
receptor. The PTB domain is colored in rainbow order from its
N-terminus (blue) to its C-terminus (red) and the target peptide
is magenta.The target peptide’s phosphoTyr (pY) side chain and
the side chain of Arg 67, which form a salt bridge (dashed line),
are drawn in stick form with C green, N blue, O red, and P
orange. Note that the pY side chain extends from a  turn on the
peptide ligand. [Based on an NMR structure by Stephen Fesik,
Abbott Laboratories,Abbott Park, Illinois. PDBid 1SHC.]
N-terminal Pro side chain (left) is largely obscured in this view
and the C-terminal three residues are disordered]. (b) The
molecular surface of the protein only, as viewed toward the
peptide binding site and colored according to its local
electrostatic potential with the most positive regions deep blue
and the most negative regions deep red.The binding pockets for
the phosphoTyr (right) and Ile (left) side chains are circled in
yellow and important residues are identified by red arrows.
[Courtesy of John Kuriyan, The Rockefeller University.]
JWCL281_c19_671-743.qxd 6/11/10 6:57 AM Page 707
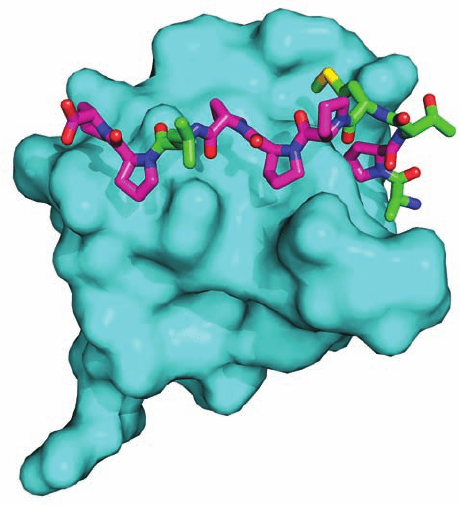
in effect, forms an additional antiparallel strand of one
of the sheets. The NPQpY segment forms a turn in
which the phosphate group contacts an Arg side chain that
mutational studies indicate is essential for the binding of
target peptide.
d. SH3 Domains Bind to Proline-Rich Peptides
Many of the RTKs that contain SH2 domains also have
one or more 50- to 75-residue SH3 domains. Moreover,
SH3 is contained in several membrane-associated proteins
that lack an SH2. The SH3 domain, which is unrelated to
SH2, binds Pro-rich sequences of 9 or 10 residues contain-
ing the motif Pro-X-X-Pro, with the residues surrounding
this motif targeting these sequences to specific SH3 do-
mains. The physiological function of SH3 is less apparent
than that of SH2 because SH3 occurs in a greater variety of
proteins, including receptor and nonreceptor tyrosine ki-
nases, adaptor proteins such as Grb2 (see below), and
structural proteins such as spectrin and myosin. However,
the observation that the deletion of the SH3 domain–
encoding segments from the proto-oncogenes Src and Abl
(which both encode PTKs) converts them to oncogenes
suggests that SH3, much like SH2 and PTB, functions to
mediate the interactions between kinases and regulatory
proteins. SH2, PTB, and SH3 have therefore been called
“molecular velcro.”
The X-ray and NMR structures of SH3 domains from
several proteins indicate that the SH3 core consists of two
3-stranded, antiparallel sheets that pack against each
other with their strands nearly perpendicular.As with SH2,
the close proximity of SH3’s N- and C-termini suggests that
this domain could be modularly inserted between two
residues on the surface of another protein without greatly
perturbing either structure. The X-ray structures of the
SH3 domains from the tyrosine kinases Abl and Fyn in
complex with two different 10-residue Pro-rich polypep-
tides to which they tightly bind were determined by
Andrea Musacchio and Matti Saraste. Both decapeptides
assume nearly identical conformations with their C-terminal
7 residues in the polyproline II helix conformation (Sec-
tion 8-1Bb). The peptides bind to SH3 over their entire
length in three geometrically complementary cavities
(Fig. 19-35), which are mostly occupied by Pro side chains.
e. Other Binding Modules
Several other binding modules have been implicated in
mediating signal transduction.These include:
1. The WW domain (named after its two highly con-
served Trp residues), an ⬃40-residue module that binds to
Pro-rich sequences on its target proteins.
2. The pleckstrin homology (PH) domain (so named
because was first recognized in pleckstrin [for platelet and
leukocyte C kinase substrate protein)], an ⬃120-residue
module that is present in 100 proteins. It structurally re-
sembles the PTB domain (Fig. 19-34) but binds to the inos-
itol head groups of phosphoinositides (Section 19-4). It
therefore targets its attached proteins to the inner surface
of the plasma membrane. In addition to its role in intracel-
lular signaling, the PH domain participates in cytoskeletal
organization, regulation of intracellular membrane trans-
port, and modification of membrane lipids. Its structure is
discussed in Section 19-4Bb.
3. The PDZ domain (named after the three proteins in
which it was first described: PSD-95, Dlg, and ZO-1), an
⬃100-residue module that mainly binds to the C-terminal
tripeptide, Ser/Thr-X-Val, of its target proteins.
Many of the proteins that participate in signal transduction
consist of several modular units that also occur in several, if
not many, other such proteins. These modules may have en-
zymatic activities (e.g., PTK activity) or bind to specific
molecular motifs, such as a phosphoTyr residue within a
specific sequence (e.g., SH2 domains) or another protein
module (e.g., SH3 domains). Apparently, signaling proteins
have arisen through the evolutionary shuffling of these
modules to generate different combinations of interactions
and activities. Indeed, we shall see that the complex behav-
ior of these signaling proteins is a consequence of the inter-
actions among their various modules.
f. Ras Is Activated by Phosphorylated RTKs
via a Grb2–Sos Complex
c-Ras (or just Ras), a proto-oncogene product, is a
monomeric membrane-anchored (by prenylation) G protein
708 Chapter 19. Signal Transduction
Figure 19-35 X-ray structure of the SH3 domain from
Abl protein in complex with its 10-residue target Pro-rich
polypeptide (APTMPPPLPP). The protein is represented by
its surface diagram (cyan) and the peptide is drawn in stick form
with Pro C magenta, other C green, N blue, O red, and S yellow.
[Based on an X-ray structure by Andrea Musacchio, European
Molecular Biology Laboratory, Heidelberg, Germany. PDBid
1ABO.]
JWCL281_c19_671-743.qxd 3/16/10 7:16 PM Page 708
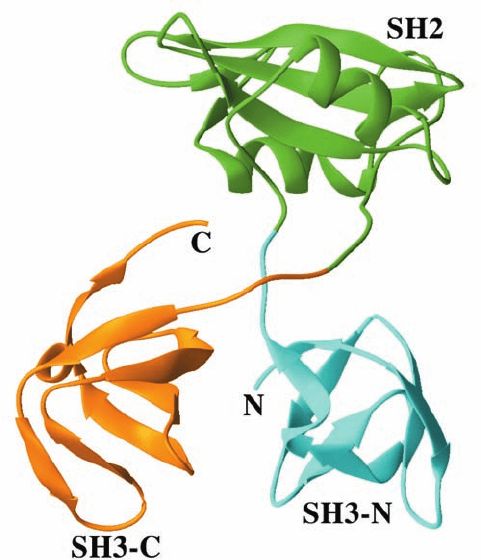
that lies at the center of an intracellular signaling system: It
regulates such essential cellular functions as growth and dif-
ferentiation through the phosphorylation and hence activa-
tion of a variety of proteins. Ras is the prototypic member
of the superfamily of small G proteins, whose 154 members
form five principal families, those of (1) Ras; (2) Arf and
(3) Rab, which both participate in vesicle trafficking (Sec-
tions 12-4Cd and 12-4Db); (4) Rho, which is mainly in-
volved in regulating the cytoskeleton (Section 35-3Ed);
and (5) Ran, which regulates nuclear import and export.
In the signaling pathway described in Section 19-3D, the
binding of ligand to RTKs ultimately activates a guanine
nucleotide exchange factor (GEF; Section 19-2Ca) to
exchange a Ras-bound GDP for GTP. Only Ras GTP
is capable of further relaying the signal. However, as do
the homologous and structurally similar subunits of the
heterotrimeric G proteins, Ras eventually hydrolyzes its
bound GTP to GDP, thereby halting further signal trans-
duction and limiting the magnitude of the signal generated
by the binding of ligand to the receptor. Indeed, the struc-
ture of Ras closely resembles those of the GTPase domains
of the heterotrimeric G protein G
subunits, including their
Switch I and Switch II regions (Fig. 19-18). Mammalian
cells express four Ras homologs: H-Ras, N-Ras, K-Ras 4A,
and K-Ras 4B.
Molecular genetic analyses of signaling in a variety of
distantly related organisms (notably humans, mice, Xeno-
pus, Drosophila, and the nematode worm Caenorhabditis
elegans) have revealed a remarkably conserved pathway in
which RTKs funnel the signal that they have bound ligand
to Ras, which, in turn, relays the signal, via a so-called MAP
kinase cascade, to the transcriptional apparatus in the nu-
cleus (Section 19-3D). Nevertheless, the way in which mes-
sages are passed between the RTKs and Ras remained
enigmatic for several years until investigations in numer-
ous laboratories revealed their major details. In particular,
these studies demonstrated that two previously character-
ized proteins, Grb2 and Sos, form a complex that bridges
activated RTKs and Ras in a way that induces Ras to ex-
change its bound GDP for GTP, thereby activating it (i.e.,
they act as a GEF).
The mammalian protein Grb2, a 217-residue homolog
of drk in Drosophila and Sem-5 in C. elegans, consists al-
most entirely of an SH2 domain flanked by two SH3 do-
mains. Sos protein (the 1596-residue product of the Son of
Sevenless gene, so named because Sos interacts with the
Sevenless gene product, an RTK that regulates the devel-
opment of the R7 photoreceptor cell in the Drosophila
compound eye), which is required for Ras-mediated signal-
ing, contains a central domain homologous to known Ras-
GEFs and a Pro-rich sequence in its C-terminal segment
similar to known SH3-binding motifs. Moreover, mam-
malian homologs of Sos (mSos) have been shown to
specifically stimulate guanine nucleotide exchange in
mammalian Ras proteins. Western blotting techniques
(Section 6-4Bc) using anti-Grb2 and anti-mSos antibodies
indicated that Grb2 binds to the C-terminal segment of
mSos but not when one of the Pro residues in Sos’ SH3-
binding motif has been replaced by Leu or in the presence
of synthetic polypeptides that have these Pro-rich se-
quences. Similar studies indicated that in the presence of
epidermal growth factor (EGF), the EGF receptor (an
RTK; Figs. 19-25 and 19-27b) specifically binds the
Grb2–mSos complex. However, this interaction is blocked
by the presence of a phosphopeptide with the sequence of
the peptide segment containing one of the activated EGF
receptor’s phosphoTyr residues. Evidently, Grb2’s SH2
domain binds a phosphoTyr-containing peptide segment in
an activated RTK while its two SH3 domains bind the Pro-
rich sequences of Sos. The GEF function of Sos is thereby
stimulated to activate Ras.
g. Grb2, Shc, and IRS Are Adaptors That
Recruit Sos to the Vicinity of Ras
The X-ray structure of Grb2, determined by Arnaud
Ducruix, reveals that neither of its SH3 domains contacts
its SH2 domain (Fig. 19-36). Moreover, although the two
SH3 domains are in contact, their interface area is rela-
tively small and hence is more likely to be an artifact of
crystallization than a structural feature of Grb2 in solution.
It therefore appears that Grb2’s SH3 domains are flexibly
linked to its SH2 domain. How does the binding of such a
pliable adaptor (a linker that lacks enzymatic activity) to a
phosphorylated RTK stimulate Sos to act as Ras’s GEF?
Grb2 and Sos bind one another so tightly that they are es-
sentially permanently associated in the cell. Hence, when
Section 19-3. Tyrosine Kinase–Based Signaling 709
Figure 19-36 X-ray structure of Grb2. Its SH2 domain (green)
is linked to its flanking SH3 domains (cyan and orange) via
apparently unstructured and hence flexible 4-residue linkers.
[Based on an X-ray structure by Arnaud Ducruix, Université de
Paris-Sud, Gif sur Yvette Cedex, France. PDBid 1GRI.]
JWCL281_c19_671-743.qxd 3/16/10 7:16 PM Page 709

Grb2 binds its target phosphorylated RTK, it recruits Sos
to the inner surface of the plasma membrane, where Sos’s
then increased local concentration causes it to more read-
ily bind to the membrane-anchored Ras.
Shc proteins are also adaptors that link activated RTKs
to Ras. Shc proteins consist of an N-terminal PTB domain
(Fig.19-34),a central effector region (CH1),and a C-terminal
SH2 domain that binds to certain activated RTKs. More-
over, Shc proteins are also major targets of various RTKs,
which phosphorylate them within their CH1 domains at
sequences that then form binding sites for the Grb2 SH2
domain. Activated RTKs may therefore bind Grb2 indi-
rectly via Shc as well as directly. Moreover, in some cases,
an Shc–Grb2–Sos complex that lacks a bound RTK may
activate Ras.
The activated (autophosphorylated) insulin receptor
does not directly interact with SH2 domain–containing
proteins. Rather, it mainly phosphorylates an ⬃1300-
residue protein named the insulin receptor substrate (IRS;
actually a family of four homologous proteins named
IRS1–4, each of which is expressed in a tissue-specific man-
ner). The IRS proteins all have an N-terminal “targeting”
region consisting of a PH domain that localizes the IRS to
the inner surface of the plasma membrane, followed by a
PTB domain that binds the IRS to a phosphoTyr residue of
an activated insulin receptor (Fig. 19-37).The insulin recep-
tor thereupon phosphorylates the IRS at one or more of its
6 to 8 Tyr residues, converting them to SH2-binding sites
that then couple this system to SH2-containing proteins
(Section 19-4F). Adaptors with multiple SH2-binding sites
such as the IRS proteins and Shc are also known as dock-
ing proteins because they function as platforms for the re-
cruitment of a variety of downstream signaling molecules
in response to the activation of their corresponding RTK.
Thus, a docking protein increases the complexity and regu-
latory flexibility of its RTK-initiated signaling pathway as
well as amplifying its signal.
h. Sos Functions to Pry Open Ras’s
Nucleotide Binding Site
The X-ray structure, determined by Kuriyan, of Ras in
complex with a 506-residue GEF-containing segment of
Sos reveals how Sos induces Ras to exchange its normally
tightly bound GDP for GTP. This Sos segment consists
of two helical domains, of which only the C-terminal
so-called catalytic domain contacts Ras. Ras is packed
against the center of the elongated bowl-shaped catalytic
domain (Fig. 19-38). Those portions of Ras that interact
with the catalytic domain include both its Switch I and
Switch II regions as well as the loop that binds the and
phosphates of GDP and GTP, the so-called P-loop (al-
though GMP binds to Ras with 10
6
-fold less affinity than
does GDP, so that the phosphate of GDP is largely re-
sponsible for its tight binding to Ras). This interaction dis-
places Switch I relative to its position in the X-ray structure
of Ras in complex with the nonhydrolyzable GTP analog
GMPPNP:
Ras’s nucleotide binding site is thereby partially opened up
and a Leu and a Glu side chain from Sos are, respectively,
introduced into Ras’ Mg
2
-binding site and the site that
binds the phosphate group of GDP/GTP. However, this
interaction does not significantly occlude Ras’s guanine
and ribose binding sites. This rationalizes how the Sos–Ras
interaction can be strong enough to displace the normally
tightly bound GDP from Ras but yet weak enough so that
H
H
O
–
CH
2
Guanosine-5-(,-imido)triphosphate
(GMPPNP)
G
O
H
H
OH OH
O
PO
O
–
O
P
O
–
–
O
O
P NH
710 Chapter 19. Signal Transduction
Figure 19-37 Structure of an insulin receptor substrate (IRS).
An IRS contains a PH and a PTB domain at its N-terminus
followed by multiple phosphoTyr-containing binding sites for the
SH2 domains of downstream signaling proteins.
PH PTB
PPPP PP P
Figure 19-38 X-ray structure of the complex between Ras and
the GEF-containing region of Sos. The N-terminal domain of the
Sos region is blue, its catalytic domain is green, and Ras is mainly
gray, with its Switch I and II regions orange and its P loop red.
Conserved regions (the SCRs) among the Ras family of GEFs
are cyan. [Courtesy of John Kuriyan, The Rockefeller University.
PDBid 1BDK.]
JWCL281_c19_671-743.qxd 3/16/10 7:17 PM Page 710