Voet D., Voet Ju.G. Biochemistry
Подождите немного. Документ загружается.

d. Structure-Based Drug Design
Since the mid 1980s, dramatic advances in the speed and
precision with which a macromolecular structure can be
determined by X-ray crystallography and NMR (Section 8-
3A) have enabled structure-based drug design, a process
that greatly reduces the number of compounds that need
be synthesized in a drug discovery program. As its name
implies, structure-based drug design (also called rational
drug design) uses the structure of a receptor in complex
with a drug candidate to guide the development of more
efficacious compounds. Such a structure will reveal, for ex-
ample, the positions of the hydrogen bonding donors and
acceptors in a receptor binding site as well as cavities in the
binding site into which substituents might be placed on a
drug candidate to increase its binding affinity for the recep-
tor. These direct visualization techniques are usually sup-
plemented with molecular modeling tools such as the com-
putation of the minimum energy conformation of a
proposed derivative, quantum mechanical calculations that
determine its charge distribution and hence how it would
interact electrostatically with the receptor, and docking
simulations in which an inhibitor candidate is computa-
tionally modeled into the binding site on the receptor to as-
sess potential interactions. Structure-based drug design is
an iterative process: The structure of the receptor in com-
plex with a compound with improved properties is deter-
mined in an effort to further improve its properties.
e. Combinatorial Chemistry, Fragment-Based Lead
Discovery, and High-Throughput Screening
As structure-based methods were developed, it ap-
peared that they would become the dominant mode of drug
discovery. However, the advent of combinatorial chemistry
techniques to rapidly and inexpensively synthesize large
numbers of related compounds combined with the develop-
ment of robotic high-throughput screening techniques has
caused the drug discovery “pendulum” to again swing to-
ward the “make-many-compounds-and-see-what-they-do”
approach.A familiar example of combinatorial chemistry is
the parallel synthesis of the large number of different
oligonucleotides on a DNA microarray (Section 7-6B). If a
lead compound can similarly be synthesized in a stepwise
manner from several smaller modules, then the substituents
on each of these modules can be varied in parallel to pro-
duce a library of related compounds (e.g., Fig. 15-30).
A variety of synthetic techniques have been developed
that permit the combinatorial synthesis of thousands of re-
lated compounds in a single procedure.Thus, whereas investi-
gations into the importance of a hydrophobic group at a par-
ticular position in a lead compound might previously have
prompted the individual syntheses of only the ethyl, propyl,
and benzyl derivatives of the compound, the use of combina-
torial synthesis would permit the generation of perhaps 100
different groups at that position. This would far more effec-
tively map out the potential range of the substituent and pos-
sibly identify an unexpectedly active analog. Interestingly,
QSAR and computational techniques have been combined in
the development of “virtual combinatorial chemistry,” a pro-
cedure in which libraries of compounds are computationally
“synthesized”and “analyzed”to predict their efficacy,thereby
again reducing the number of compounds that must actually
be synthesized in order to generate an effective drug.
The relatively recently developed approach to drug dis-
covery known as fragment-based lead discovery (FBLD)
has shown encouraging signs of success. In this method,
rather than screening large numbers of potential lead com-
pounds for substances that bind with high affinity to a drug
target, only a relatively small number of simple compounds
are screened for their ability to bind to the drug target with
low affinity. Substances that do so, which likely bind to only
a small portion of the drug target’s surface area, are then
Section 15-4. Drug Design 541
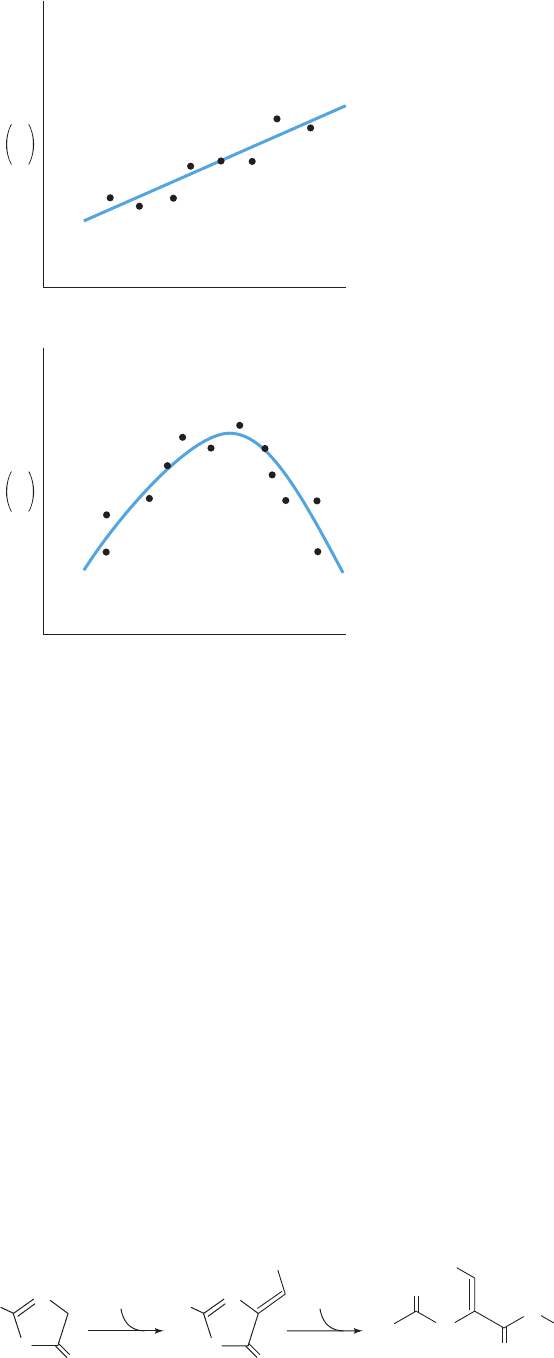
Figure 15-29 Hypothetical QSAR plots of log(1/C) versus log
P for a series of related compounds. (a) A plot that is best
described by a linear equation. (b) A plot that is best described
by a quadratic equation.
Figure 15-30 The combinatorial synthesis of
arylidene diamides. If ten different variants of each
R group are used in the synthesis, then 1000
different derivatives will be synthesized.
1
__
C
log
1
__
C
log
logP
logP
(a)
(b)
N
O
O
R
1
N
O
O
R
1
R
1
R
3
N
H
R
2
CHO
R
2
R
3
NH
2
H
N
O
O
R
2
JWCL281_c15_506-556.qxd 2/19/10 9:28 PM Page 541
“grown” by adding chemical groups and/or linking several
such fragments together. Thus, FBLD discovers a lead
compound one piece at a time rather than all at once.
B. Introduction to Pharmacology
The in vitro development of an effective drug candidate is
only the first step in the drug development process. Besides
causing the desired response in its isolated target receptor, a
useful drug must be delivered in sufficiently high concentra-
tion to this receptor where it resides in the human body with-
out causing unacceptable side effects.
a. Pharmacokinetics Is a Multifaceted Phenomenon
The most convenient form of drug administration is
orally (by mouth). In order to reach its target receptor, a
drug administered in this way must surmount a series of for-
midable barriers: (1) It must be chemically stable in the
highly acidic (pH 1) environment of the stomach and must
not be degraded by the digestive enzymes in the gastroin-
testinal tract; (2) it must be absorbed from the gastrointesti-
nal tract into the bloodstream, that is, it must pass through
several cell membranes; (3) it must not bind too tightly to
other substances in the body (e.g., lipophilic substances
tend to be absorbed by certain plasma proteins and by fat
tissue; anions may be bound by plasma proteins, mainly al-
bumin; and cations may be bound by nucleic acids); (4) it
must survive derivatization by the battery of enzymes,
mainly in the liver, that function to detoxify xenobiotics
(foreign compounds), as discussed below (note that the in-
testinal blood flow drains directly into the liver via the por-
tal vein, so that the liver processes all orally ingested sub-
stances before they reach the rest of the body); (5) it must
avoid rapid excretion by the kidneys; (6) it must pass from
the capillaries to its target tissue; (7) if it is targeted to the
brain, it must cross the blood–brain barrier, which blocks
the passage of most polar substances; and (8) if it is targeted
to an intracellular receptor, it must pass through the plasma
membrane and, possibly, other intracellular membranes.
The ways in which a drug interacts with these various barri-
ers is known as its pharmacokinetics. Thus, the bioavailabil-
ity of a drug (the extent to which it reaches its site of action,
which is usually taken to be the systemic circulation) de-
pends on both the dose given and its pharmacokinetics. Of
course, barriers (1) and (2) can be circumvented by inject-
ing the drug [e.g., some forms of penicillin (Fig. 11-28) must
be injected because their functionally essential -lactam
rings are highly susceptible to acid hydrolysis], but this
mode of drug delivery is undesirable for long-term use.
Since the pharmacokinetics of a drug candidate is as im-
portant to its efficacy as is its pharmacodynamics, both
must be optimized in producing a medicinally useful drug.
The following empirically based rules, formulated by
Christopher Lipinski and known as Lipinski’s “rule of
five,” state that an orally administered compound is likely
to exhibit poor absorption or permeation if:
1. Its molecular mass is greater than 500 D.
2. It has more than 5 hydrogen bond donors (expressed
as the sum of its OH and NH groups).
3. It has more than 10 hydrogen bond acceptors (ex-
pressed as the sum of its N and O atoms).
4. Its value of log P is greater than 5.
Drug candidates that disobey Rule 1 are likely to have low
solubilities and to only pass through cell membranes with
difficulty; those that disobey Rules 2 and/or 3 are likely to be
too polar to pass through cell membranes;and those that dis-
obey Rule 4 are likely to be poorly soluble in aqueous solu-
tion and hence unable to gain access to membrane surfaces.
Thus, the most effective drugs are usually a compromise; they
are neither too lipophilic nor too hydrophilic. In addition,
their pK values are usually in the range 6 to 8 so that they
can readily assume both their ionized and unionized forms
at physiological pH’s. This permits them to cross cell mem-
branes in their unionized form and to bind to their receptor
in their ionized form. However, since the concentration of a
drug at its receptor depends, as we saw, on many different
factors, the pharmacokinetics of a drug candidate may be
greatly affected by even small chemical changes. QSARs
and other computational tools have been developed to pre-
dict these effects but they are, as yet, rather crude.
b. Toxicity and Adverse Reactions Eliminate Most
Drug Candidates
The final criteria that a drug candidate must meet are that
its use be safe and efficacious in humans. Tests for these
properties are initially carried out in animals, but since hu-
mans and animals often react quite differently to a particu-
lar drug, the drug must ultimately be tested in humans
through clinical trials. In the United States, clinical trials are
monitored by the Food and Drug Administration (FDA)
and have three increasingly detailed (and expensive) phases:
Phase I. This phase is primarily designed to test the safety
of a drug candidate but is also used to determine its dosage
range and the optimal dosage method (e.g., orally vs injec-
tion) and frequency. It is usually carried out on a small
number (20–100) of normal, healthy volunteers, but in the
case of a drug candidate known to be highly toxic (e.g., a
cancer chemotherapeutic agent), it is carried out on volun-
teer patients with the target disease.
Phase II. This phase mainly tests the efficacy of the drug
against the target disease in 100 to 500 volunteer patients
but also refines the dosage range and checks for side effects.
The effects of the drug candidate are usually assessed via
single blind tests, procedures in which the patient is un-
aware of whether he/she has received the drug or a control
substance. Usually the control substance is a placebo (an in-
ert substance with the same physical appearance, taste, etc.,
as the drug being tested) but, in the case of a life-threaten-
ing disease, it is an ethical necessity that the control sub-
stance be the best available treatment against the disease.
Phase III. This phase monitors adverse reactions from long-
term use as well as confirming efficacy in 1000 to 5000 pa-
tients. It pits the drug candidate against control substances
through the statistical analysis of carefully designed double
blind tests, procedures in which neither the patients nor the
clinical investigators evaluating the patients’ responses to
the drug know whether a given patient has received the
542 Chapter 15. Enzymatic Catalysis
JWCL281_c15_506-556.qxd 2/19/10 9:28 PM Page 542

drug or a control substance.This is done to minimize bias in
the subjective judgments the investigators must make.
Currently, only about 5 drug candidates in 5000 that en-
ter preclinical trials reach clinical trials. Of these, only one,
on average, is ultimately approved for clinical use, with
⬃40% of drug candidates passing Phase I trials and ⬃50%
of those passing Phase II trials (most drug candidates that
enter Phase III trials are successful). In recent years, the
preclinical portion of a drug discovery process has aver-
aged ⬃3 years to complete, whereas successful clinical tri-
als have usually required an additional 7 to 10 years. These
successive stages of the drug discovery process are increas-
ingly expensive, so that to successfully bring a drug to mar-
ket costs, on average, around $300 million.
The most time-consuming and expensive aspect of a
drug development program is identifying a drug candi-
date’s rare adverse reactions. Nevertheless, it is not an un-
common experience for a drug to be brought to market
only to be withdrawn some months or years later when it is
found to have caused unanticipated life-threatening side
effects in as few as 1 in 10,000 individuals (the search for
new applications of an approved drug and its postmarket-
ing surveillance are known as its Phase IV clinical trials).
For example, in 1997, the FDA withdrew its approval of the
drug fenfluramine (fen),
among individuals as well as differences in their disease
states, other drugs they are taking, age, sex, and environmen-
tal factors. The cytochromes P450, which function in large
part to detoxify xenobiotics and participate in the meta-
bolic clearance of the majority of drugs in use, provide in-
structive examples of these phenomena.
The cytochromes P450 constitute a superfamily of heme-
containing enzymes that occur in nearly all living organisms,
from bacteria to mammals [their name arises from the char-
acteristic 450-nm peak in their absorption spectra when re-
acted in their Fe(II) state with CO].The human genome en-
codes 57 isozymes (catalytically and structurally similar but
genetically distinct enzymes from the same organism; also
called isoforms) of cytochromes P450, about one-third of
which occur only in the liver (P450 isozymes are named by
the letters “CYP” followed by a number designating its fam-
ily, an uppercase letter designating its subfamily, and often
another number; e.g., CYP2D6). These monooxygenases
(Fig.15-31), which in animals are embedded in the endoplas-
mic reticulum membrane, catalyze reactions of the sort
The electrons (e
) are supplied by NADPH, which passes
them to cytochrome P450’s heme prosthetic group via the
intermediacy of the enzyme NADPH–P450 reductase.
Here RH represents a wide variety of usually lipophilic
compounds for which the different cytochromes P450 are
specific. They include polycyclic aromatic hydrocarbons
[PAHs, frequently carcinogenic (cancer-causing) com-
pounds that are present in tobacco smoke, broiled meats,
and other pyrolysis products], polycyclic biphenyls (PCBs,
RH O
2
2H
2e
Δ ROH H
2
O
Section 15-4. Drug Design 543
CH
3
C NH
2
CH
2
CH
3
CF
3
Fenfluramine Phentermine
CH
2
CH
2
CH
3
CH
3
CH NH
Figure 15-31 X-ray structure of the human cytochrome P450
CYP2C9 in complex with the blood clotting inhibitor warfarin. A
cutaway diagram of the enzyme’s active site region drawn with
its surface purple and with its polypeptide backbone in ribbon
form colored in rainbow order from N-terminus (blue) to
C-terminus (red).The heme (seen edgewise), the Cys side chain
that axially ligands the heme Fe atom, and the warfarin
(coumadin; Fig. 35-12) are shown in ball-and-stick form with C
gray, N blue, O red, S yellow, and Fe orange. [Courtesy of Astex
Therapeutics, Limited. PDBid 1OG5.]
which it had approved in 1973 for use as an appetite sup-
pressant in short-term (a few weeks) weight loss programs.
Fenfluramine had become widely prescribed, often for ex-
tended periods, together with another appetite suppressant,
phentermine (phen; approved in 1959), a combination
known as fen-phen (although the FDA had not approved of
the use of the two drugs in combination, once it approves a
drug for any purpose, a physician may prescribe it for any
other purpose, a practice known as off-label use).The with-
drawal of fenfluramine was prompted by over 100 reports
of heart valve damage in individuals (mostly women) who
had taken fen-phen for an average of 12 months (phenter-
mine was not withdrawn because the evidence indicated
that fenfluramine was the responsible agent).This rare side
effect had not been observed in the clinical trials of fenflu-
ramine, in part because, being an extremely unusual type of
drug reaction, it had not been screened for.
More recently (2004), the widely prescribed analgesic
Vioxx was withdrawn from use due to its previously unde-
tected cardiac side effects, although the closely related
analgesic Celebrex remains available (Section 25-7Bb).
c. The Cytochromes P450 Metabolize Most Drugs
Why is it that a drug that is well tolerated by the major-
ity of patients can pose such a danger to others? Differ-
ences in reactions to drugs arise from genetic differences
JWCL281_c15_506-556.qxd 2/19/10 9:28 PM Page 543
which were widely used in electrical insulators and as plas-
ticizers and are also carcinogenic), steroids (in whose syn-
theses cytochromes P450 participate; Sections 25-6A and
25-6C), and many different types of drugs. The xenobiotics
are thereby converted to a more water-soluble form, which
aids in their excretion by the kidneys. Moreover, the newly
generated hydroxyl groups are often enzymatically conju-
gated (covalently linked) to polar substances such as glu-
curonic acid (Section 11-1Cb), glycine, sulfate, and acetate,
which further enhances aqueous solubility.The many types
of cytochromes P450 in animals, which have different sub-
strate specificities (although these specificities tend to be
broad and hence often overlap), are thought to have arisen
in response to the numerous toxins which plants produce,
presumably to discourage animals from eating them (other
P450s function to catalyze specific biosynthetic reactions).
Drug–drug interactions are often mediated by cy-
tochromes P450. For example,if drug A is metabolized by or
otherwise inhibits a cytochrome P450 isozyme that metabo-
lizes drug B, then coadministering drugs A and B will cause
the bioavailability of drug B to increase above the value it
would have had if it alone had been administered.This phe-
nomenon is of particular concern if drug B has a low thera-
peutic index. Conversely, if, as is often the case, drug A in-
duces the increased expression of the cytochrome P450
isozyme that metabolizes it and drug B, then co-adminis-
tering drugs A and B may reduce drug B’s bioavailability, a
phenomenon that was first noted when certain antibiotics
caused oral contraceptives to lose their efficacy. Moreover, if
drug B is metabolized to a toxic product, its increased rate of
reaction may result in an adverse reaction. Environmental
pollutants such as PAHs or PCBs are also known to induce
the expression of specific cytochrome P450 isozymes and
thereby alter the rates at which certain drugs are metabo-
lized. Finally, some of these same effects may occur in pa-
tients with liver disease, as well as arising from age-based,
gender-based, and individual differences in liver physiology.
Although the cytochromes P450 presumably evolved to
detoxify and/or help eliminate harmful substances, in sev-
eral cases they have been shown to participate in converting
relatively innocuous compounds to toxic agents. For exam-
ple, acetaminophen (Fig. 15-32), a widely used analgesic and
antipyretic (fever reducer), is quite safe when taken in ther-
apeutic doses (1.2 g/day for an adult) but in large doses
(10 g) is highly toxic. This is because, in therapeutic
amounts, 95% of the acetaminophen present is enzymati-
cally glucuronidated or sulfated at its group to the
corresponding conjugates, which are readily excreted. The
remaining 5% is converted, through the action of a cy-
tochrome P450 (CYP2E1), to acetimidoquinone (Fig. 15-32),
which is then conjugated with glutathione, a tripeptide with
an unusual -amide bond that participates in a wide variety
of metabolic processes (Section 26-4C). However, when
acetaminophen is taken in large amounts, the glucuronida-
¬OH
544 Chapter 15. Enzymatic Catalysis
Figure 15-32 The metabolic reactions
of acetaminophen that convert it to its
conjugate with glutathione.
N
H
C
CH
3
O
OH
S
OH
HO
OH
O
cytochrome P450
O
2
H
2
O
spontaneous
H
2
O
Acetaminophen
(N-acetyl-p-aminophenol)
Acetimidoquinone
Glutathione
(␥-L-Glutamyl-L-cysteinyl-glycine)
H
3
NCH CH
SH
CH
2
CH
2
CH
2
CH
2
COO
–
COO
–
CCNH NH
O
+
O
Acetaminophen–glutathione conjugate
H
3
NCH CHCH
2
CH
2
CH
2
CH
2
COO
–
COO
–
CCNH NH
O
+
N
C
CH
3
O
N
C
CH
3
O
O
N
H
C
CH
3
O
JWCL281_c15_506-556.qxd 2/19/10 9:28 PM Page 544
tion and sulfation pathways become saturated and hence
the cytochrome P450-mediated pathway becomes increas-
ingly important. If hepatic (liver) glutathione is depleted
faster than it can be replaced, acetimidoquinone, a reactive
compound, instead conjugates with the sulfhydryl groups of
cellular proteins, resulting in often fatal hepatotoxicity.
Many of the cytochromes P450 in humans are unusually
polymorphic, that is, there are several common alleles
(variants) of the genes encoding these enzymes. Alleles
that cause diminished, enhanced, and qualitatively altered
rates of drug metabolism have been characterized for
many of the cytochromes P450. The distributions of these
various alleles differ markedly among ethnic groups and
hence probably arose to permit each group to cope with
the toxins in its particular diet.
Polymorphism in a given cytochrome P450 results in dif-
ferences between individuals in the rates at which they me-
tabolize certain drugs. For instance, in cases that a cy-
tochrome P450 variant has absent or diminished activity,
otherwise standard doses of a drug that the enzyme nor-
mally metabolizes may cause the bioavailability of the drug
to reach toxic levels. Conversely, if a particular P450 en-
zyme has enhanced activity (usually because the gene en-
coding it has been duplicated one or more times), higher
than normal doses of a drug that the enzyme metabolizes
would have to be administered to obtain the required ther-
apeutic effect. However, if the drug is metabolized to a
toxic product, this may result in an adverse reaction. Sev-
eral known P450 variants have altered substrate specifici-
ties and hence produce unusual metabolites, which also
may cause harmful side effects.
Experience has amply demonstrated that there is no
such thing as a drug that is entirely free of adverse reactions.
However, as the enzymes and their variants that partici-
pate in drug metabolism are characterized and rapid and
inexpensive genotyping methods are developed, it may be-
come possible to tailor drug treatment to an individual’s
genetic makeup rather than to the population as a whole.
This rapidly developing area of study is known as pharma-
cogenomics (or, more colloquially, as “personalized ge-
nomics” or “personalized medicine”).
C. HIV Protease and Its Inhibitors
Acquired immunodeficiency syndrome (AIDS), the only
major epidemic attributable to a previously unknown
pathogen to appear in the 20th century (it was first de-
scribed in 1981), is caused by human immunodeficiency
virus type 1 (HIV-1; the closely related HIV-2, which we
shall not explicitly discuss here, also causes AIDS and has a
similar response to drugs). HIV-1, which was discovered in
1983 by Françoise Barré-Sinoussi, Luc Montagnier, and
Robert Gallo, is a retrovirus, a family of viruses that were
independently characterized in 1970 by David Baltimore
and Howard Temin. The retroviral genome is a single-
stranded RNA that reproduces inside its host cell by tran-
scribing the RNA to double-stranded DNA in a process
mediated by the virally encoded enzyme reverse transcrip-
tase (Section 30-4C). The DNA is then inserted into the
host cell’s chromosomal DNA by a viral enzyme named in-
tegrase and is passively replicated along with the cell’s
DNA. However, under activating conditions (which for
HIV-1 often is an infection by another pathogen), the retro-
viral DNA is transcribed, the proteins it encodes are ex-
pressed and inserted in or anchored to the host cell plasma
membrane, and new virions (virus particles) are produced
by the budding out of a viral protein–laden segment of
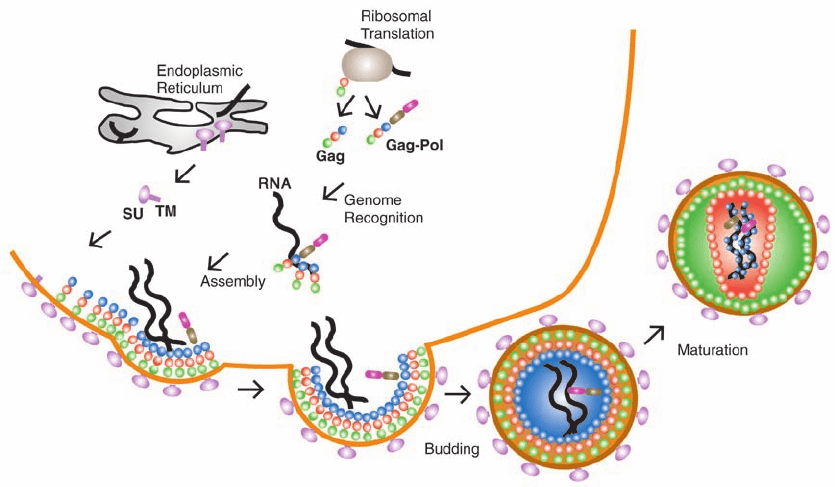
plasma membrane so as to enclose viral RNA (Fig. 15-33).
HIV-1 is targeted to and specifically replicates within
helper T cells, essential components of the immune system
Section 15-4. Drug Design 545
Figure 15-33 The assembly, budding, and maturation of HIV-1. SU is the surface glycoprotein gp120 and
TM is the transmembrane protein gp41. [After Turner, B.G. and Summers, M.F., J. Mol. Biol. 285, 4 (1999).]
JWCL281_c15_506-556.qxd 2/19/10 9:28 PM Page 545
(Section 35-2Aa). Unlike most types of retroviruses, HIV-1
eventually kills the cells producing it. Although the helper T
cells within which HIV-1 are actively replicating are often de-
stroyed by the immune system, those within which the HIV-
1 is latent (its DNA is not being transcribed) are not detected
by the immune system and hence provide a reservoir of HIV-
1 (other types of cells also harbor HIV-1).Consequently, over
a several year period after the initial infection (during most
of which the host exhibits no obvious symptoms), the host’s
immune system is steadily depleted until it has deteriorated
to the point that the host regularly falls victim to and is even-
tually killed by opportunistic pathogens that individuals with
normally functioning immune systems can readily withstand.
It is this latter stage of an HIV infection that is called AIDS.
In the absence of effective therapy,AIDS is almost invariably
fatal. Through the year 2008, nearly 30 million people had
died of AIDS and an estimated 35 million others, largely in
sub-Saharan Africa, were HIV-positive, numbers that are in-
creasing at the rate of ⬃3 million per year.As a consequence
of this global catastrophe, HIV has been characterized and
effective countermeasures against it have been devised faster
than for any other pathogen in history.
a. Reverse Transcriptase Inhibitors
Are Only Partially Effective
The first drug to be approved by the FDA (in 1987) to fight
AIDS was 3ⴕ-azido-3ⴕ-deoxythymidine (AZT; zidovudine),
H
H
H
H
H
NNN
O
HOCH
2
T
3
ⴕ-Azido-3ⴕ-deoxythymidine
(AZT; zidovudine)
+
⫺
_
which had first been synthesized in 1964 as a possible anti-
cancer agent (it was ineffective). AZT is a nucleoside ana-
log that, on enzymatic conversion to its triphosphate in the
cell (the plasma membrane is impermeable to nucleoside
triphosphates), inhibits HIV-1 reverse transcriptase, as do
the several other drugs (Section 30-4Ca) that the FDA had
approved to treat AIDS prior to 1996. Unfortunately, these
agents only slow the progression of an HIV infection but
do not stop it.This is in part because they are toxic, mainly
to the bone marrow cells that are blood cell precursors, and
hence cannot be taken in large doses. More important,
however, is that reverse transcriptase, unlike most other
DNA polymerases (Section 30-2A), cannot correct its mis-
takes and hence frequently generates mutations (about
one per 10
4
bp and, since the viral genome consists of ⬃10
4
bp, each viral genome bears, on average, one new muta-
tion). Consequently, under the selective pressure of an anti-
HIV drug such as AZT, the drug’s target receptor rapidly
evolves to a drug-resistant form.
b. HIV-1 Polyproteins Are Cleaved by HIV-1 Protease
HIV-1, as do other retroviruses, synthesizes its proteins in
the form of polyproteins, which each consist of several
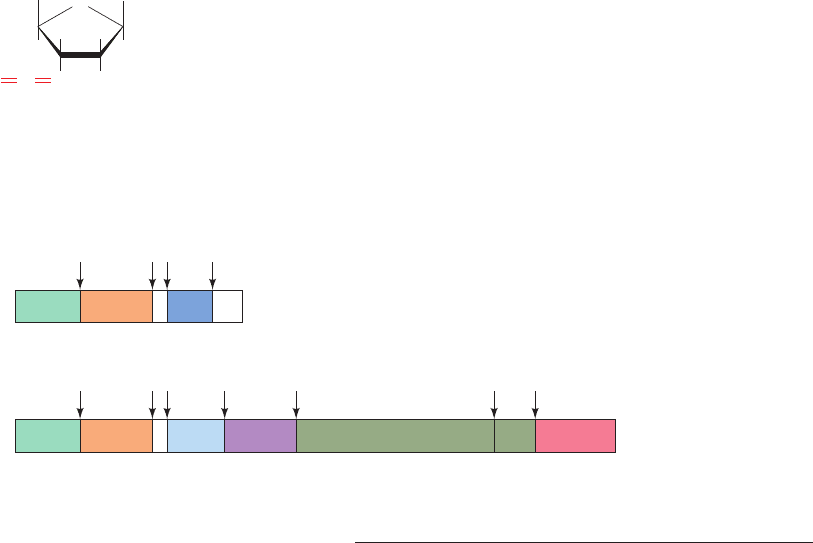
tandemly linked proteins (Fig. 15-34). HIV-1 encodes two
polyproteins, gag (55 kD) and gag–pol (160 kD), which are
both anchored to the plasma membrane via N-terminal
myristoylation (Section 12-3Bb).These polyproteins are then
cleaved to their component proteins through the action of
HIV-1 protease, but only after this enzyme has excised itself
from gag–pol. This process occurs only after the virion has
budded off from the host cell and results in a large structural
reorganization of the virion (Fig. 15-33).The virion is thereby
converted from its noninfectious immature form to its patho-
genic mature form. If HIV-1 protease is inactivated, either
mutagenically or by an inhibitor,the virion remains noninfec-
tious. Hence HIV-1 protease is an opportune drug target.
c. Aspartic Proteases and Their Catalytic Mechanism
HIV-1 protease is a member of the aspartic protease fam-
ily (also known as acid proteases), so called because these en-
zymes all contain catalytically essential Asp residues that
546 Chapter 15. Enzymatic Catalysis
Figure 15-34 HIV-1 polyproteins. (a) The organization of the
HIV-1 gag and gag–pol polyproteins. The symbols used are MA,
matrix protein; CA, capsid protein; NC, nucleocapsid protein;TF,
transmembrane protein; PR, protease; RT, reverse transcriptase;
RN, ribonuclease; and IN, integrase. (b) The sequences flanking
the HIV-1 protease cleavage sites (red bonds) indicated in Part a.
TF
p1
CAMA PR RT RN IN
I II III V VI VII VIII
gag–pol
NC
p1
CAMA p6
I II III IV
gag
(a)
Cleavage Sequence
site
I
II
III
IV
V
VI
VII
VIII
...
Ser -Gln-Asn- Tyr — Pro - Ile - Val -Gln
...
...
Ala-Arg- Val -Leu — Ala-Glu-Ala-Met
...
...
Ala- Thr - Ile -Met — Met-Gln-Arg- Gly
...
...
Pro -Gly-Asn-Phe — Leu-Gln- Ser -Arg
...
...
Ser -Phe-Asn-Phe — Pro -Gln- Ile - Thr
...
...
Thr -Leu-Asn-Phe — Pro - Ile - Ser - Pro
...
...
Ala-Glu- Thr-Phe — Tyr - Val -Asp-Gly
...
...
Arg- Lys - Ile -Leu — Phe-Leu-Asp-Gly
...
(b)
JWCL281_c15_506-556.qxd 2/19/10 9:28 PM Page 546
occur in the signature sequence Asp–Thr/Ser–Gly. Humans
have several known aspartic proteases including pepsin, a
digestive enzyme secreted by the stomach (its specificity is
indicated in Table 7-2) that functions at pH 1 and which was
the first enzyme to be recognized (named in 1836 by
Theodor Schwann); chymosin (formerly rennin), a stomach
enzyme, occurring mainly in infants, that specifically cleaves
a Phe–Met peptide bond in the milk protein -casein,
thereby causing milk to curdle, making it easier to digest
(calf stomach chymosin has been used for millennia to make
cheese); cathepsins D and E, lysosomal proteases that func-
tion to degrade cellular proteins; renin, which participates
in the regulation of blood pressure and electrolyte balance
(Fig. 15-35); and -secretase (also known as memapsin 2), a
transmembrane protein common in brain that participates
in cleaving A precursor protein to yield amyloid- protein
(A), which is implicated in Alzheimer’s disease (Section 9-
5B). In addition, many fungi secrete aspartic proteases, pre-
sumably to aid them in invading the tissues they colonize.
Eukaryotic aspartic proteases are ⬃330-residue mono-
meric proteins. The X-ray structure of pepsin (Fig. 15-36a),
which closely resembles those of other eukaryotic aspartic
proteases, reveals that this croissant-shaped protein con-
sists of two homologous domains that are related by ap-
proximate 2-fold symmetry (although only about 25
residues in the core sheets of each domain are closely re-
lated by this symmetry). Each domain contains a catalyti-
cally essential Asp in an analogous position. The X-ray
structures of enzyme–inhibitor complexes of various aspar-
tate proteases indicate that substrates bind in a prominent
cleft between the two domains that could accommodate an
⬃8-residue polypeptide segment in an extended
sheet–like conformation. The active site Asp residues are
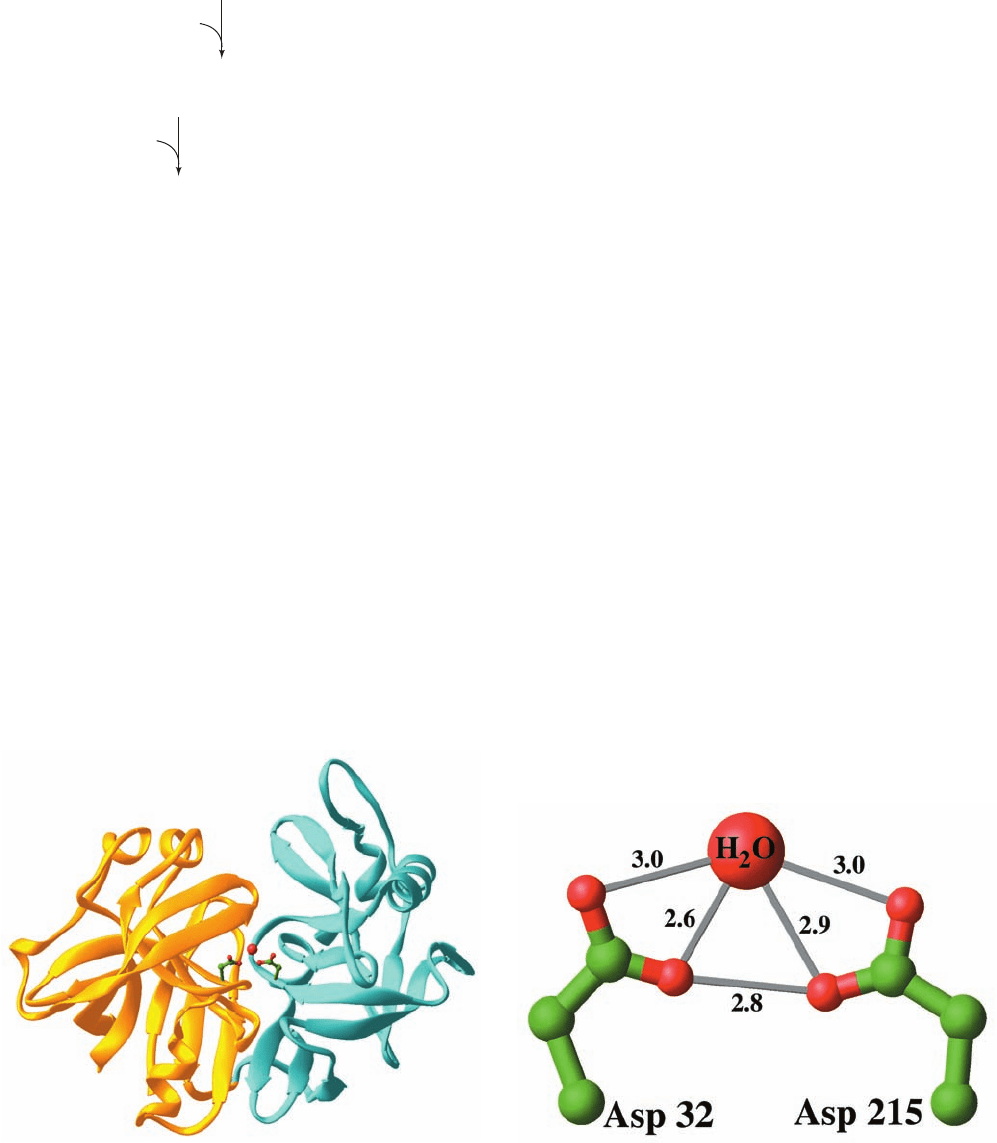
located at the base of this cleft (Fig. 15-36a).
What is the catalytic mechanism of eukaryotic aspartic
proteases? Proteolytic enzymes, in general, have three es-
sential catalytic components:
1. A nucleophile to attack the carbonyl C atom of the
scissile peptide to form a tetrahedral intermediate (Ser 195
serves this function in trypsin; Fig. 15-23).
2. An electrophile to stabilize the negative charge that
develops on the carbonyl O atom of the tetrahedral inter-
mediate (the H bonding donors lining the oxyanion hole,
Gly 193 and Ser 195, do so in trypsin; Fig. 15-25).
Section 15-4. Drug Design 547
Figure 15-35 Renin participation in blood pressure regulation.
Renin proteolytically cleaves the 13-residue polypeptide
angiotensinogen to the 10-residue polypeptide angiotensin I.
This latter peptide is then cleaved by angiotensin converting
enzyme (ACE) to the 8-residue polypeptide angiotensin II,
which, on binding to its receptor, induces vasoconstriction and
retention of Na
and water by the kidneys, resulting in increased
blood pressure. Consequently several inhibitors of renin and of
ACE have been developed for the control of hypertension (high
blood pressure).
Figure 15-36 X-ray structure of porcine pepsin. (a) Ribbon
diagram in which the N-terminal domain (residues 1–172) is
gold, the C-terminal domain (residues 173–326) is cyan, the side
chains of the active site Asp residues are shown in ball-and-stick
form with C green and O red, and the water molecule that is
bound by these Asp side chains is represented by a red sphere.
The protein is viewed with the pseudo-2-fold axis relating core
1
Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu-Val-Ile-His
13
Angiotensinogen
H
2
O renin
1
Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu
10
+ Val-Ile-His
H
2
O angiotensin converting enzyme (ACE)
Angiotensin I
1
Asp-Arg-Val-Tyr-Ile-His-Pro-Phe
8
+ His-Leu
Angiotensin II
portions of the two domains tipped from vertical toward the
viewer. (b) Enlarged view of the active site Asp residues and
their bound water molecule indicating the lengths (in Å) of
possible hydrogen bonds (thin gray bonds).The X-ray structures
of other aspartic proteases exhibit similar interatomic distances.
[Based on an X-ray structure by Anita Sielecki and Michael
James, University of Alberta, Edmonton, Canada. PDBid 4PEP.]
(a)
(b)
JWCL281_c15_506-556.qxd 2/19/10 9:28 PM Page 547
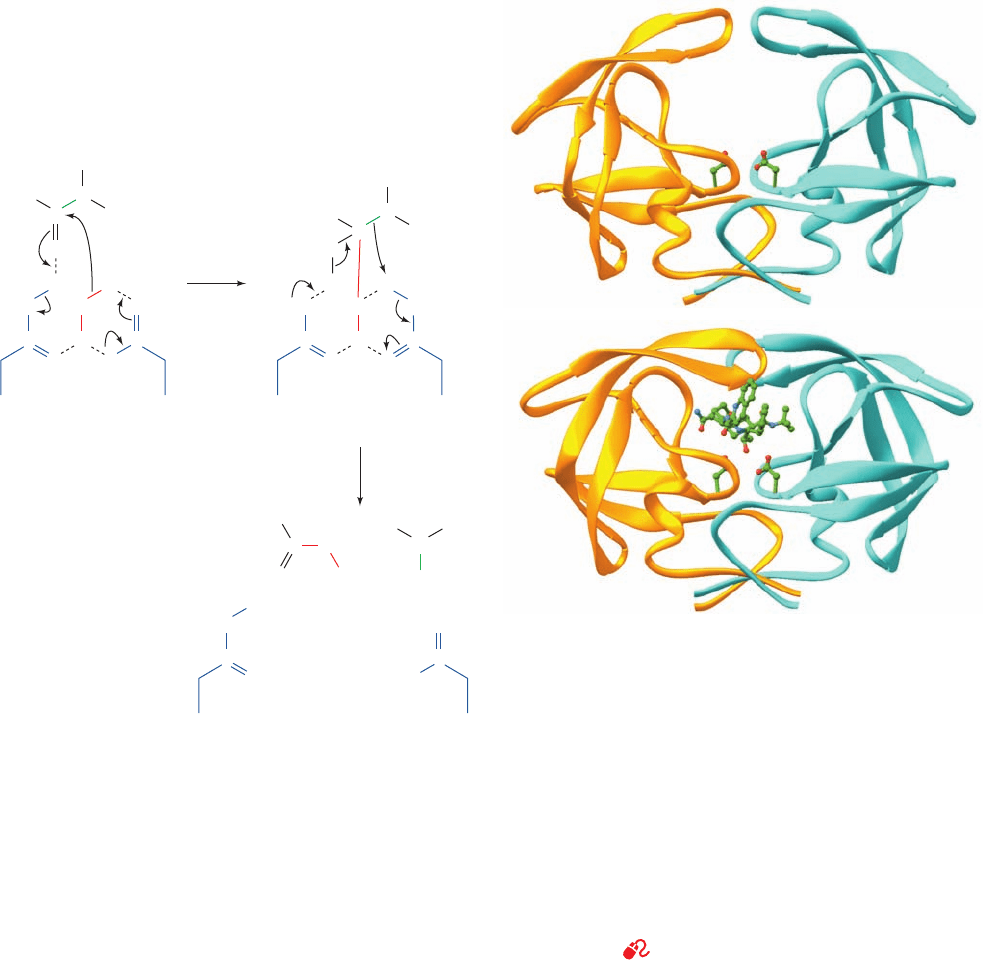
3. A proton donor so as to make the amide N atom of
the scissile peptide a good leaving group (the imidazolium
group of His 57 in trypsin; Fig. 15-23).
Pepsin’s pH rate profile (Section 14-4) suggests that it
has two ionizable essential residues, one with pK ⬇ 1.1 and
the other with pK ⬇ 4.7,which are almost certainly the car-
boxyl groups of its essential Asp residues. At the pH of the
stomach, the Asp residue with pK 4.7 is protonated and
that with pK 1.1 is partially ionized. This suggests that the
ionized carboxyl group acts as a nucleophile to form the
putative tetrahedral intermediate. However, no covalent
intermediate between an aspartic protease and its sub-
strate has ever been detected.
The two active site Asp residues in eukaryotic aspartic
proteases are in close proximity and both appear to form
hydrogen bonds to a bridging water molecule that is pres-
ent in several X-ray structures of eukaryotic aspartic pro-
teases (Fig. 15-36b). This, together with a variety of enzy-
mological and kinetic data, led Thomas Meek to propose
the following catalytic mechanism for aspartic proteases
(Fig. 15-37):
1. An active site Asp carboxylate group,acting as a gen-
eral base, activates the bound water molecule, the so-called
lytic water, to nucleophilically attack the scissile peptide’s
carbonyl C as an OH
⫺
ion. Proton donation (general acid
catalysis) by the second, previously uncharged active site
Asp stabilizes the oxyanion that would otherwise form in
the resulting tetrahedral intermediate.
2. The N atom of the scissile peptide is protonated by
the first Asp (general acid catalysis) resulting, through
charge rearrangement and proton transfer to the second
Asp (general base catalysis), in amide bond scission.
Aspartic proteases are inhibited by compounds with tetra-
hedral carbon atoms at a position mimicking a scissile pep-
tide bond (see below). This strongly suggests that these
548 Chapter 15. Enzymatic Catalysis
Figure 15-37 Catalytic mechanism of aspartic proteases. (1)
The nucleophilic attack of the enzyme-activated water molecule
(red) on the carbonyl carbon atom of the scissile peptide bond
(green) to form the tetrahedral intermediate. This reaction step is
promoted by general base catalysis by the Asp on the right and
general acid catalysis by the Asp on the left (blue). (2) The
decomposition of the tetrahedral intermediate to form products
via general acid catalysis by the Asp on the right and general
base catalysis by the Asp on the left.
CC
C
H
H
O
O
O
O
Asp Asp
Michaelis complex
H
O O
R
R⬘
N
H
–
CC
C
H
H
O
O
O
O
Asp Asp
Tetrahedral intermediate
H
O O
R
R⬘
N
H
–
–
C
C
H
O
O
O
Asp Asp
Products
O
O
H
O
+
C
R
R⬘
N
H
H
1
2
Figure 15-38 X-ray structure of HIV-1 protease.
(a) Uncomplexed and (b) in complex with its inhibitor
saquinavir (structural formula in Fig. 15-41). In each structure,
the homodimeric protein is viewed with its 2-fold axis of
symmetry vertical and is shown as a ribbon diagram with one
subunit gold and the other cyan.The side chains of the active site
Asp residues,Asp 25 and Asp 25¿, as well as the saquinavir in
Part b, are shown in ball-and-stick form with C green, N blue,
and O red. Note how the  hairpin “flaps” at the top of the
uncomplexed enzyme have folded down over the inhibitor in the
saquinavir complex. Compare these structures with that of the
similarly viewed pepsin in Fig. 15-36a. [Part a based on an X-ray
structure by Tom Blundell, Birkbeck College, London, U.K., and
Part b based on an X-ray structure by Robert Crowther,
Hoffmann-LaRoche Ltd., Nutley, New Jersey. PDBids (a) 3PHV
and (b) 1HXB.]
See Interactive Exercise 7
(a)
(b)
JWCL281_c15_506-556.qxd 10/19/10 7:20 AM Page 548
enzymes preferentially bind their transition states (transi-
tion state stabilization), thereby enhancing catalysis.
d. HIV-1 Protease Inhibitors Are Effective
Anti-AIDS Agents
HIV-1 protease differs from eukaryotic aspartic pro-
teases in that it is a homodimer of 99-residue subunits. Nev-
ertheless, its X-ray structure (Fig. 15-38a), determined inde-
pendently in 1989 by Alexander Wlodawer, by Manuel
Navia and Paula Fitzgerald, and by Tom Blundell, closely
resembles those of eukaryotic aspartic proteases. Thus,
HIV-1 protease has the enzymatically unusual property that
its single active site is formed by two identical symmetri-
cally arranged subunits. Quite possibly HIV-1 protease re-
sembles the putative primordial aspartic protease that,
through gene duplication, evolved to form the eukaryotic
enzymes (although HIV-1 protease is well suited to the lim-
ited amount of genetic information that a virus can carry).
Once the structure of HIV-1 protease became available,
intensive efforts were mounted in numerous laboratories
to find therapeutically effective inhibitors of this enzyme.
In this process, nearly 300 X-ray structures and several
NMR structures have been reported of HIV-1 protease, its
mutants, and the proteases of other retroviruses, both
alone and in their complexes with a great variety of in-
hibitors. Hence, HIV-1 protease is perhaps the most ex-
haustively structurally studied protein.
Comparison of the X-ray structure of HIV-1 protease
alone (Fig. 15-38a) with that of its complexes with
polypeptide-like inhibitors (e.g., Fig. 15-38b) reveals that,
on binding an inhibitor, the hairpin “flaps” covering the
“top” of the substrate-binding cleft swing down by as much
as 7 Å to enclose the inhibitor. Such an inhibitor binds to
the 2-fold symmetric enzyme in a 2-fold pseudosymmetric
extended conformation such that the inhibitor interacts
with the enzyme much like a strand in a sheet (Fig. 15-39).
On the “floor” of the binding cleft, each signature sequence
(Asp 25–Thr 26–Gly 27) is located in a loop that is stabi-
lized by a network of hydrogen bonds similar to that ob-
served in eukaryotic aspartic proteases.The inhibitor inter-
acts with the enzyme via a hydrogen bond to the active site
residue Asp 25. However, contrary to the case for eukary-
otic aspartic proteases (Fig. 15-36b), no X-ray structure
of an HIV-1 protease contains a water molecule within
Section 15-4. Drug Design 549
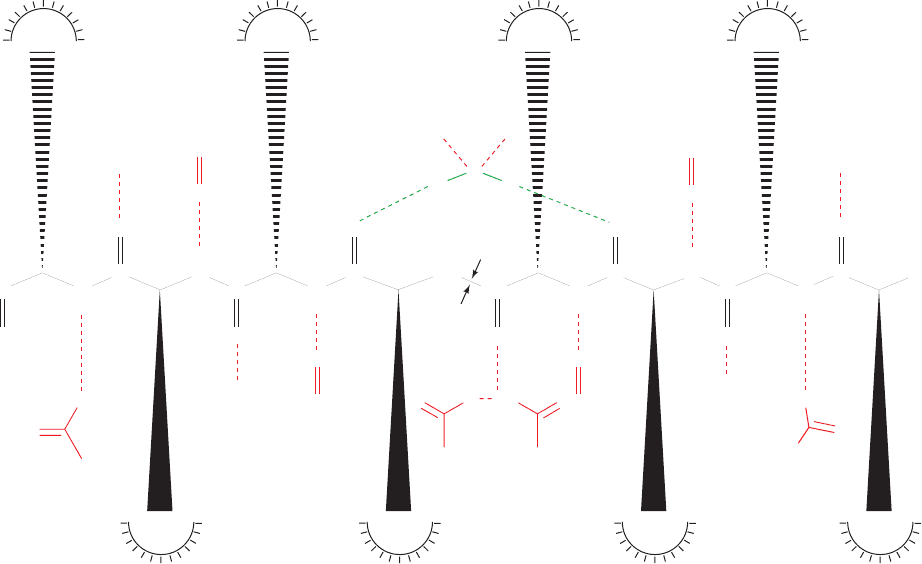
Figure 15-39 Arrangement of hydrogen bonds between HIV-1
protease and a modeled substrate. In the nomenclature used
here, polypeptide residues in one subunit are assigned primed
numbers to differentiate them from the residues of the other
subunit; substrate residues on the N-terminal side of the scissile
peptide bond are designated P
1
,P
2
,P
3
, … , counting toward the
P
4
⬘
S
4
⬘
P
3
⬘
S
3
⬘
P
1
⬘
S
1
⬘
P
2
S
2
P
4
S
4
P
1
S
1
P
3
S
3
C
C
C
N
H
N
H
H
N
H
N
O
O
O
O
C
C
C
N
H
N
H
N
H
O
O
O
C
O
C
O
N
H
Asp 29⬘
Asp 29⬘
Ile 50⬘ Ile 50
Asp 29
Gly 27⬘
Gly 48⬘
H
N
H
N
C
C
N
H
O
O
O
Gly 27
Gly 48 Gly 48
H
N
H
N
CN
H
O
O
O
⫺
O
C
HH
Flap side
P
2
⬘
S
2
⬘
O
⫺
Asp 25
HO
O
Asp 25⬘
⫺
O
O
N-terminus; substrate residues on its C-terminal side are
designated P¿
1
,P¿
2
,P¿
3
, …, counting toward the C-terminus; and the
symbols S
1
,S
2
,S
3
, …, and S¿
1
,S¿
2
,S¿
3
, …, designate the enzyme’s
corresponding residue-binding subsites.The scissile peptide bond
is marked by arrows. [After Wlodawer,A. and Vondrasek, J.,
Annu. Rev. Biophys. Biomol. Struct. 27, 257 (1998).]
JWCL281_c15_506-556.qxd 6/5/10 9:10 AM Page 549
hydrogen bonding distance of Asp 25 or Asp 25¿. On the
flap side of the binding cleft, the inhibitor interacts with
Gly 48 and Gly 48¿ and with a water molecule that is not
the attacking nucleophile but which mediates the contacts
between the flaps and the inhibitor backbone.
Although HIV-1 protease specifically cleaves the gag
and gag–pol polyproteins at a total of 8 sites (Fig. 15-34b),
these sites appear to have little in common except that
their immediately flanking residues are nonpolar and
mostly bulky. Indeed, binding studies indicate that HIV-1
protease’s specificity arises from the cumulative effects of
the interactions between the enzyme and the amino acids
in positions P
4
through P¿
4
. However, three of the peptides
cleaved by HIV-1 have either the sequence Phe-Pro or Tyr-
Pro, which are sequences that human aspartic proteases do
not cleave. Hence, HIV-1 protease inhibitors containing
groups that resemble either of these dipeptides would be
unlikely to inhibit essential human aspartic proteases.
An effective HIV-1 protease inhibitor should resemble a
substrate with its scissile peptide replaced by a group that the
enzyme cannot cleave. Such a group should, preferably, en-
hance the enzyme’s affinity for the inhibitor. Mimics of the
tetrahedral intermediate (Fig. 15-37), that is, transition state
analogs, are likely to do so. Consequently, a variety of such
groups (Fig. 15-40) have been investigated in efforts to syn-
thesize therapeutically effective inhibitors of HIV-1 protease.
Although HIV-1 protease has high in vitro affinity for
its polypeptide-based inhibitors, these substances have
poor oral bioavailability (they are degraded by digestive
550 Chapter 15. Enzymatic Catalysis
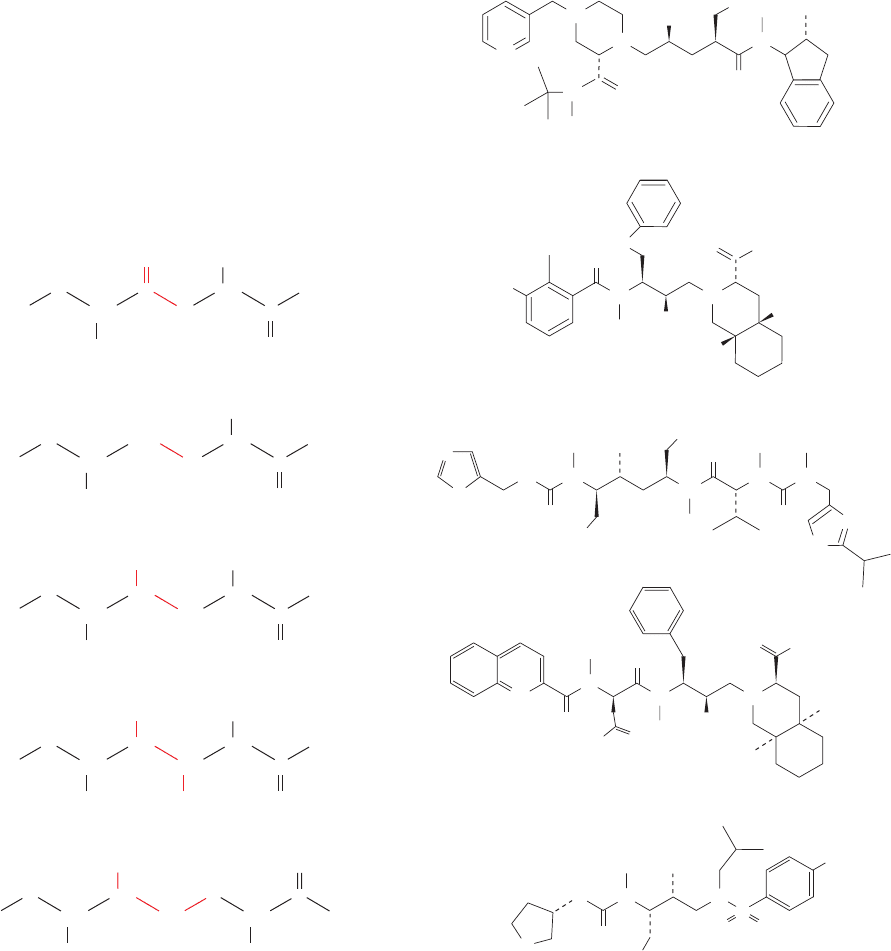
Figure 15-40 Comparison of a normal peptide bond (top) to a
selection of groups (red) that are isosteres (stereochemical
analogs) of the tetrahedral intermediate in reactions catalyzed by
aspartic proteases.
Figure 15-41 Some HIV-1 protease inhibitors that are in
clinical use. Note that in addition to its generic (chemical) name,
each drug has a proprietary trade name, here in parentheses,
under which it is marketed.
Indinavir (Crixivan
TM
)
Nelfinavir (Viracept
TM
)
Ritonavir (Norvir
TM
)
Saquinavir (Invirase
TM
)
Amprenavir (Agenerase
TM
)
N
N
NN
N
O
OH
O
H
OH
•H
2
SO
4
H
Ph
•CH
3
SO
3
H
HO
O
N
N
H
S
OH
O
NHtBu
H
H
S
N
N
O
O
O
O
H
N
H
N
H
OH
Ph
Ph
N
S
•CH
3
SO
3
H
N
N
O
O
O
O
H
N
H
NH
2
OH
N
NHtBu
H
H
NH
2
O
O
O
N
N
H
Ph
OH
S
O
O
CH
3
N
H
N
N
H
C
CH
CHC
O
C
O
O
R⬘
Peptide bond
H
N
N
H
C
CH
CHCH
2
O
R⬘
R
R
Reduced amide
H
N
CH
2
C
CH
CHCH
OH
O
R⬘
R
Hydroxyethylene
H
N
CH
C
CH
CHCH
OH
O
R⬘
R⬘
R
OH
Dihydroxyethylene
H
N
CH
2
CH
CH
H
NCH
OH
R
Hydroxyethylamine
JWCL281_c15_506-556.qxd 6/7/10 2:05 PM Page 550