Messerschmidt U. Dislocation Dynamics During Plastic Deformation
Подождите немного. Документ загружается.

80 4 Dislocation Motion
x
d
b
u
x
y
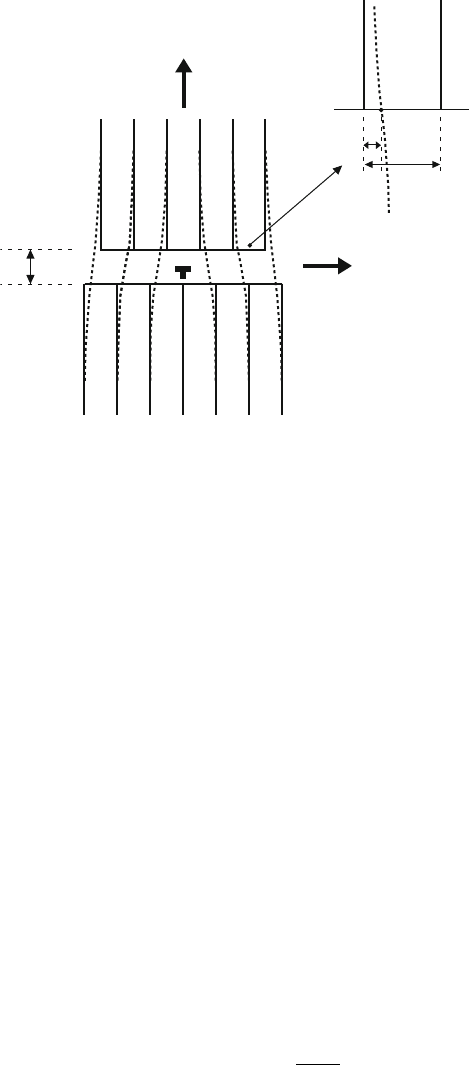
Fig. 4.3. Joining two semi-infinite elastically strained crystals to form an edge
dislocation
by springs and moving on a periodic substrate. At the same time, Orowan
suggested the problem of dislocation motion in a periodic potential relief to
Peierls who solved it in analytical form [127]. Later on it was extended by
Nabarro [128].
In the Peierls–Nabarro model, an edge dislocation is simulated by two
elastic half-crystals, which are shifted with respect to each other by half the
Burgers vector b as outlined in Fig. 4.3 by the two sets of parallel lattice planes.
The half-crystals are positioned at a distance d. To join the lattice planes of the
two half-crystals, these planes have to be displaced on the glide plane in the
middle between the two half-crystals (y =0)by
2u
x
(x)+b/2forx>0
2u
x
(x) −b/2forx<0
with respect to the bottom half-crystal. The displaced and joined lattice planes
are represented by dotted lines in Fig. 4.3. Far from the dislocation, the dis-
placements have to fulfil the boundary condition u
x
(∞)=−u
x
(−∞)=−b/4.
To realize the displacements, internal stresses have to be applied. Similar
to the Frenkel treatment of the theoretical shear strength of Sect. 1.1, it
is assumed that the stresses depend on the displacements via a sinusoidal
function and that the displacements in y direction are comparatively small
σ
xy
(x, 0) = −K sin
4πu
x
b
.

4.2 Lattice Friction 81
K is a constant, which is determined by fulfilling Hooke’s law for small strains
μ =
1
2
∂σ
xy
∂ε
xy
=
d
2
∂σ
xy
∂u
x
,
where ε
xy
(x, 0) = (2u
x
+ b/2)/(2d). For u
x
= 0, it follows that
σ
xy
(x, 0) = −
μb
2πd
sin
4πu
x
b
. (4.16)
As found out by Eshelby [129], this shear stress distribution can be realized
by a continuous distribution of infinitesimal edge dislocations along the x axis,
leading to an integral equation with the solution
u
x
= −
b
2π
arctan
2x(1 − ν)
d
. (4.17)
This relation fulfils the boundary condition stated above. Besides, u
x
(d/[2(1−
ν)]) = −b/8=(1/2)u
x
(∞). Accordingly, in the range −d/[2(1 − ν)] <x<
d/[2(1 −ν)], the disregistry is greater than half its maximum value of −b/4at
x = ∞. This is a possibility to define a width of the dislocation within which
linear elasticity theory is certainly not valid. The width replaces the core
region of the Volterra dislocation, which is cut out by the inner cut-off radius.
The method of a continuous distribution of infinitesimal edge dislocations
also allows the calculation of the whole stress field of the Peierls dislocation.
The relations reduce to the stress field of the Volterra dislocation (3.11) for
distances great with respect to d/[2(1 − ν)]. The Peierls dislocation does not
show the divergence of the stresses in the core of the Volterra dislocation.
The energy of the Peierls dislocation is composed of the elastic energy of
the two half-crystals and the misfit energy of the atom rows between them.
For comparing the result with that of the Volterra dislocation, the elastic
energy can be calculated in two half-cylinders, which have to be deformed by
surface forces to yield the displacements u
x
. Following the original treatments
of Peierls [127] and Nabarro [128], the result is
W (x)=
μb
2
4π(1 − ν)
+
μb
2
2π(1 − ν)
exp −
2πd
b(1 − ν)
cos
4πx
b
=
μb
2
4π(1 − ν)
+
W
p
2
cos
4πx
b
. (4.18)
W
p
is called the Peierls energy and represents the magnitude of the periodic
displacement potential. The dislocation energy varies with double the lattice
periodicity b because of the two symmetrical positions in Fig. 4.2. However,
(4.18) does not correspond to the expectation from Fig. 4.2 as the symmetrical
positions turn out to be positions of maximum energy. This result originates
from the method of summing up the energies of the individual lattice rows in
the two original treatments. The method was improved by Hirth and Lothe

82 4 Dislocation Motion
[130] omitting this problem. The maximum of the derivative of W (x)with
respect to the position x of the dislocation yields the stress necessary to move
the dislocation over the potential barrier
τ
p
=
1
b
∂W(x)
∂x
max
=
2πW
p
b
2
=
2μ
1 − ν
exp −
2πd
b(1 − ν)
. (4.19)
This is the Peierls–Nabarro stress or shortly Peierls stress to move a
straight dislocation lying in a Peierls valley only by mechanical stress, that
is, at very low temperatures. For d ≈ b, it is in the range of 10
−4
...10
−2
μ,
that means in the range of the real shear strengths of materials. The lattice
resistance is controlled by the structure of the dislocation cores. Therefore,
the Peierls–Nabarro model is too coarse for detailed predictions of the flow
stress of crystals. Similar to the theoretical yield stress of perfect crystals in
Sect. 1.1, the Peierls stress is small for small Burgers vectors b and large dis-
tances d between the lattice planes. According to the experimental results,
the Peierls stress is small for close-packed structures like f.c.c. metals, where
it does not play a role except at low temperatures. It is high for materials
with directional bonds like the covalent semiconductor and ceramic crystals,
which become plastic only at high temperatures. The b.c.c. metals show an
intermediate behavior.
The Peierls–Nabarro model is refined in many ways: by relaxing the severe
assumptions made and by using more realistic potentials than the sinusoidal
one (e.g. [131]). More detailed information on the dislocation core configura-
tions and energies as well as the Peierls stress can be obtained from atomistic
calculations. The crystal is divided into an outer zone, which is treated by
continuum elasticity theory and a core region where the interactions between
the atoms are calculated discretely. The positions that are first determined by
the elastic solution are then varied to find the equilibrium configurations. The
results of these calculations depend very sensitively on the chosen interaction
potentials between the atoms. The situation is comparatively simple in ionic
crystals with Coulomb attraction, Born–Mayer repulsion, and van-der-Waals
potentials. Early calculations of this kind were made in [132] for NaCl and
in [133] for MgO. For metallic materials, the semi-empirical embedded-atom
method (EAM) is quite successful (for a review see [134]). In recent studies
of dislocation cores and the Peierls stress, details of the dislocation behavior
were interpreted, for example, for intermetallic alloys [135] or on the influence
of shear stresses in more than one slip plane and of nonglide stresses in b.c.c.
Mo [136]. In a model cubic primitive lattice, the Peierls stress was calculated
by molecular dynamics simulation [137]. Applying a sinusoidal interaction
between the atoms, the Peierls potential also turns out to be close to sinu-
soidal, as assumed in most continuum models. As a general result of the
atomistic calculations, it may be stated that the effective core radius intro-
duced in Sect. 3.2.2, which is chosen to include the core energy within the
elastic solution, is slightly less than 0.5b in ionic crystals, and 0.5b toafewb
in metals.

4.2 Lattice Friction 83
Hirth and Lothe [12] suggest to use a phenomenological equation for the
Peierls potential
W (x)=
W
p
2
1 − cos
2πx
h
= W
p
sin
2
πx
h
, (4.20)
where h either equals b/2orb, depending on the degree of equivalence of the
two equilibrium configurations. Accordingly, the Peierls stress becomes
τ
p
=
πW
p
bh
. (4.21)
4.2.2 Double-Kink Model
In the same way in which the high energy necessary to shear two lattice planes
with respect to each other as a whole is expended in small steps by the motion
of a straight dislocation from one Peierls valley to the next, this shift can
again be performed in small steps by the generation and sidewise spreading
of kinks. At low stresses, the kinks may occur in their thermal equilibrium
concentration. At higher stresses, they form under the action of the stress. For
dislocations not lying in a Peierls valley, that is, not being parallel to a low-
index crystallographic direction, kinks are necessary for geometric reasons. As
shown in Fig. 4.4a, kinks may appear as single kinks K or kink pairs K
+
K
−
.
Depending on the binding properties of the material, the kinks can be abrupt
as in Fig. 4.4a or may have a defined width w as in Fig. 4.4b. If the kinks
are treated within the framework of the line tension model, the dislocation
changes in a smooth way from one Peierls valley to the next (Fig. 4.4c). The
sidewise motion of the kinks along the dislocation as indicated by the arrows
in Fig. 4.4c shifts the dislocation forward (upwards) in small steps.
Elastic Properties of Kinks
The properties of the kink formation depend on their energy, which can be
estimated from continuum elasticity theory. Kink pairs or double-kinks of
height h can be formed by putting a segment of length L of a straight dislo-
cation into the neighboring Peierls valley. For kinks of small h with respect to
L, the energy of the kink pair is given by
W
fdk
=2W
fk
+ W
int
, (4.22)
where W
fk
is the formation energy of a single kink and W
int
is the interac-
tion energy between both kinks. Using the configuration of Fig. 4.4a of sharp
kinks, the kink formation energy can be determined for kinks of height h
large compared to the inner cut-off radius r
0
. This condition is not fulfilled
for elementary kinks. Since the long-range stress field of a dislocation is not
disturbed remarkably by introducing a kink, the core energy of (3.16) of a
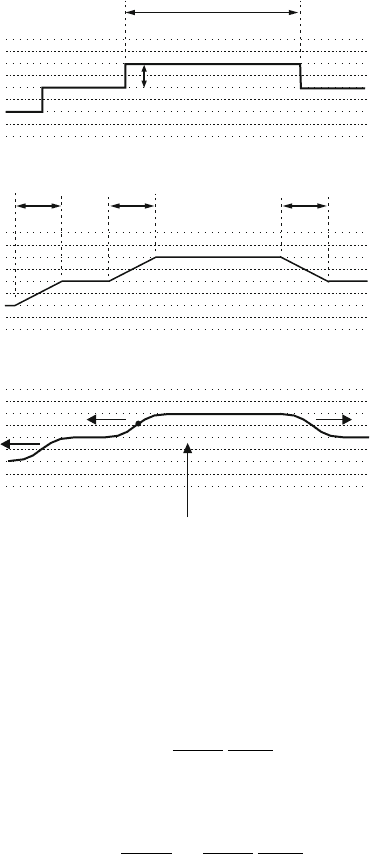
84 4 Dislocation Motion
ww w
(b)
(c)
K
K
+
K
-
(a)
L
h
x
P
Fig. 4.4. Configurations of single kinks K and kink pairs K
+
K
−
.(a) Abrupt kinks.
(b)Kinksofwidthw.(c) Smooth kinks. Dotted lines: Peierls valleys, dashed lines:
Peierls hills
piece of dislocation of length h can be taken for the kink formation energy,
W
fk
≈ (1/10)μb
2
h. This problem does not occur for the interaction energy.
For a kink pair in a screw dislocation, it is given by
W
int
= −
μb
2
h
2
8πL
1+ν
1 − ν
. (4.23)
There follows the attractive interaction force
F
int
=
∂W
int
∂L
=
μb
2
h
2
8πL
2
1+ν
1 − ν
. (4.24)
This expression can be verified by applying Brown’s theorem (3.44). The
same equation holds for the extended kink configuration in Fig. 4.4b as long
as L>w. For a kink pair in an edge dislocation, the factor 1 + ν has to
be replaced by 1 − 2ν (see Table 3.2 for the line tensions of screw and edge
dislocations). It should be noted that the interaction force decreases quickly
with 1/L
2
in contrast to long dislocations where the interaction force decreases
with the reciprocal distance (e.g. Sect. 3.2.4). The calculation of the energy

4.2 Lattice Friction 85
of individual kinks is more complicated as they have to form at a surface so
that image terms have to be considered.
The kink energy can also be determined using the line tension model in its
simple version of constant line tension equal to the line energy E
d
, for instance
(3.17). The dislocation is assumed to have the smooth kink configuration
of Fig. 4.4c. At some point P of the kink, the line tension acts as a force
in tangential direction of the dislocation line. This force has a component
in forward x direction. It is balanced at any point along the kink by the
force ∂W(x)/∂x resulting from the Peierls potential, taken, for example, from
(4.20), and by the force from the applied stress τ
∗
. Thus, the equilibrium
condition is [138]
dE
dx
= −E
d
d
2
x
dz
2
+
dW
p
dx
− τ
∗
b =0. (4.25)
The solution of the problem for zero stress yields
W
fk
=
2h
π
2W
p
E
d
. (4.26)
This formula, which expresses the kink formation energy in terms of the
Peierls energy and the dislocation line energy, links the Peierls–Nabarro model
for the motion of a straight dislocation at low temperature, that is, a quantity
describing the periodic lattice potential, with the double-kink model of realis-
tic dislocation motion. For the formation of the kinks at constant temperature,
the free energy of kink formation ΔF
fk
has to be used instead of the energy.
However, the uncertainties of the elastic estimates above are greater than the
entropy contribution so that ΔF
fk
≈ W
fk
. Like the dislocation energy W (x)in
(4.18),thekinkenergy,too,showsalattice periodicity resulting in an energy
barrier to the motion of kinks. The respective potential is sometimes called
the Peierls potential of second order.
Kinks in Thermal Equilibrium
At a finite temperature, the equilibrium configuration of a dislocation is not
a straight line lying in a Peierls valley but a dislocation containing a certain
concentration of kinks. Inside the crystal, the kinks can only be formed as kink
pairs with equal numbers of positive and negative kinks and a well-defined free
energy of formation 2ΔF
fk
of the kink pairs. Their concentration is governed
by a minimum in the free energy of the system, that is,
∂ΔF
∂n
.
=0, (4.27)
if n is the number of kinks of one sign along a dislocation of length L.Each
kink increases the free energy of the system by ΔF
fk
, yielding the total increase
nΔF
fk
.
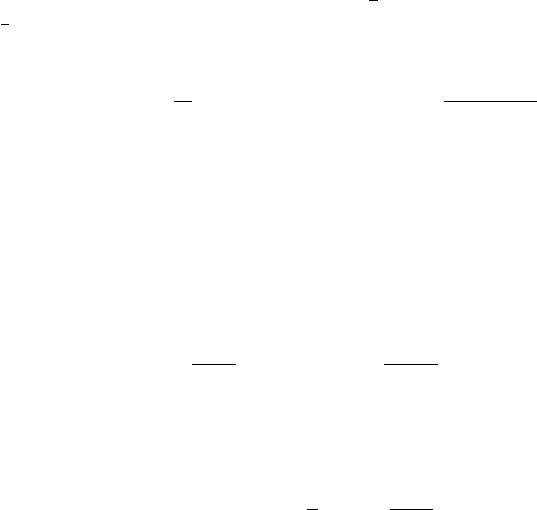
86 4 Dislocation Motion
On the other hand, a configurational entropy occurs
ΔS = k ln P
n
,
where P
n
is the number of possibilities to arrange the n kinks of one sign
at the respective number of possible kink sites N = L/a inside L.Fora
single kink, P
1
= N.Fortwokinks,P
2
=
1
2
N(N − 1). For three kinks, P
3
=
1
6
N(N − 1)(N − 2). The factors 1/2, 1/6, etc. consider that the individual
kinks cannot be distinguished from each other. Thus, for n kinks of one sign
P
n
=
1
n!
N(N − 1) ...(N − n +1)=
N!
(N − n)!n!
. (4.28)
For the change in the free energy by introducing n kink pairs, this yields
ΔF =2(nΔF
fk
− kT ln P
n
) .
Inserting (4.28) and using Stirling’s formula for large n,
ln x!=x ln x − x
yields
∂ΔF
∂n
=ΔF
fk
− kT ln
N − n
n
.
=0
for the equilibrium condition (4.27). Introducing the kink concentrations of
positive and negative kinks c
+
= c
−
= n/L and considering that n N
finally results in an equilibrium concentration of kinks
c
+
= c
−
=
1
a
exp
−
ΔF
fk
kT
. (4.29)
This derivation is typical also for a number of other processes where defects
exist in thermal equilibrium. A very prominent case, which will be discussed in
Sect. 4.10, is the occurrence of thermal point defects, vacancies, and interstitial
atoms.
Thermally Activated Motion of Kinks
The kink formation energy, which frequently is of the order of magnitude of
the dislocation core energy, is not very high, for simple metals it is only a
fraction of one electron-volt. The kink migration energy is due to the periodic
variation of the kink energy. This variation is sometimes called the secondary
Peierls potential. It is mostly small so that the kink motion can be aided
by thermal activation. Since the height of the elementary kinks is also small
(in the order of b), the force acting from the applied stress is small, too.
Therefore, the kink motion can be treated by the theory of diffusion. For a

4.2 Lattice Friction 87
general introduction into diffusion see [139, 140]. The diffusion coefficient for
the diffusion of kinks along the dislocation line is defined by
D
k
= a
2
ω, (4.30)
where a is the jump distance of the kink and ω the jump frequency. When
asmallforceF acts on the kink, the jump frequency in forward direction
becomes higher than that in backward direction, resulting in a drift velocity,
which is given by the Einstein relation
v
k
=
D
k
kT
F. (4.31)
In general, the force F can originate from a mechanical stress, but as
thermodynamic force also from a chemical potential μ = kT ln(c/c
0
), where
c is the concentration of some species and c
0
its equilibrium value. The force
is then
F = −
∂μ
∂x
= −
kT
c
∂c
∂x
. (4.32)
Equations (4.31) and (4.32) together yield Fick’s first law
J = cv
k
= −D
k
∂c
∂x
. (4.33)
Here, J is the flux or current density. The frequency ω to move a kink over
the activation barrier is given by
ω ≈ ν
0
exp
−
ΔF
mk
kT
. (4.34)
In this case, the attempt frequency ν
0
is approximately equal to the Debye
frequency ν
D
=10
11
...10
12
s
−1
.ΔF
mk
is the free energy of kink migration.
For kink motion, the force in (4.31) results from a shear stress τ acting on the
slip system of the dislocation, that is,
F = τbh.
Correctly speaking, τ is the locally acting stress. The superscript * is dropped
in the following considerations. Together with (4.30), (4.31), and (4.34), this
relation yields the velocity of the kink along the dislocation
v
k
= D
k
τbh
kT
≈ ν
D
a
2
exp
−
ΔF
mk
kT
τbh
kT
. (4.35)
Except in crystals with strong directional bonds, the kink migration energy
is quite small and much smaller than the kink formation energy. Therefore,
kinks often move very fast so that their velocity is controlled by high speed
damping processes, which will briefly be described in Sect. 4.12. Under certain

88 4 Dislocation Motion
conditions, the diffusion coefficient of kinks is then constant, approximately
equal to the limiting case of ΔF
mk
kT , but this coincidence is fortuitous.
To conclude the dislocation velocity v
d
from the kink velocity, the concen-
tration of kinks c
k
must be known
v
d
= hc
k
v
k
. (4.36)
There are different situations depending on the magnitude of the force acting
on the kinks, or on the applied stress. For small stresses, the kink concentra-
tion does not differ remarkably from the thermal equilibrium concentration
in (4.29). For high stresses, kink pairs are formed by stress-assisted thermal
activation so that the concentration of kinks is much higher than in the small
stress regime. In the latter case, (4.29) and (4.35) can be inserted into (4.36).
Considering further that c
k
= c
+
+ c
−
yields an expression for the dislocation
velocity in the small stress region,
v
d
=
2τbh
2
akT
D
k
exp
−
ΔF
fk
kT
. (4.37)
Double-Kink Nucleation at High Stresses
At high stresses, kink pairs are formed by stress-assisted thermal activation.
Only kink pairs moving the dislocation forward, which are favored by the
applied stress, need to be considered as kink pairs moving the dislocation
backwards will immediately shrink and annihilate. Because of the mutual
elastic interaction energy of the kinks of the pair (4.23), which decreases with
increasing kink separation L, and the increasing energy gained from the exter-
nal stress, the formation energy of a double-kink ΔF
fdk
will show a maximum
at a critical separation L
∗
of the kinks, as schematically shown in Fig. 4.5. At
the critical separation, the attractive kink interaction force is balanced by the
force τbh due to the external stress, which drives the kinks apart. Kink pairs
with separations smaller than L
∗
collapse with a gain in energy. Those with
L>L
∗
tend to separate further. However, kink pairs close to L
∗
can still
annihilate so that only those which are wider than L
are really stable. The
energy difference between the states at L
∗
and L
is in the order of magni-
tude of kT. This behavior of the free energy of double-kink formation and the
related nucleation and annihilation properties very much resemble the steady
state nucleation theory [141–145] as it was developed, for example, for crystal
growth [146]. With some approximations, the result of this theory yields the
double-kink nucleation rate
J
dk
=
τbh
a
2
kT
D
k
exp
−
ΔF
∗
fdk
kT
. (4.38)
ΔF
∗
fdk
is the maximum value of the double-kink formation energy at the crit-
ical separation L
∗
. To obtain the dependence of the double-kink formation

4.2 Lattice Friction 89
Fig. 4.5. Schematic plot of the dependence of the double-kink formation energy on
the separation of the kinks. Dashed line: without external stress. Solid line:with
external stress. See text below
energy ΔF
fdk
(L) on the kink separation as depicted in Fig. 4.5, both the contri-
bution of the kink interaction energy (4.23) introduced by Seeger and Schiller
[147, 148] and the work done by the external stress have to be considered.
With average data of screw and edge dislocations, the formation free energy
for kink pairs of a separation larger than the kink width w canbewrittenas
ΔF
fdk
(L)=2ΔF
fk
−
μb
2
h
2
8πL
− τbhL. (4.39)
The critical separation L
∗
of the kink pairs can be obtained by
∂ΔF
fdk
∂L
L=L
∗
=0,
yielding
L
∗
=
μbh
8πτ
.
Inserting this into (4.39) results in the critical double-kink formation energy
ΔF
∗
fdk
=2ΔF
fk
−
μτb
3
h
3
2π
. (4.40)
Evaluations of experimental data are often based on this formula, c.f. [149].
For a relatively low stress of τ =10
−4
μ and h = b, the critical kink separation
is L
∗
=20b. Thus, the kinks are well separated. Using (4.19) and (4.26), a
Peierls stress of τ
p
=10
−2
μ, and a dislocation energy of E
d
= μb
2
/2 (3.17),
the formation energy of two separated kinks becomes 2ΔF
fk
=10
√
8b
3
μ/π
3/2
.
Considering that μb
3
=5eV,2ΔF
fk
≈ 0.25 eV. Compared to this value, the
last term in (4.40) amounts to only 10% of the total energy. Thus, for this