Lennarz W.J., Lane M.D. (eds.) Encyclopedia of Biological Chemistry. Four-Volume Set . V. 4
Подождите немного. Документ загружается.

in viral infection and GCN2 by uncharged tRNAs in
amino acid starvation. Our group noted the existence of
a predicted transmembrane protein in the C. elegans
genome, with an N-terminal “extracellular” domain
similar to IRE1 and an intracellular domain similar to
PKR and GCN2, known eIF2
a
kinases. This protein,
which is conserved in metazoans (and absent from yeast)
and to which we gave the name PERK (PKR-like ER
Kinase), couples ER stress to eIF2
a
phosphorylation
and is essential to translational regulation by ER stress.
Cells lacking PERK are hypersensitive to ER stress
and in mammals, PERK activity is especially important
for preservation of cells engaged in high levels of
secretion. Thus humans with PERK mutations and
mice lacking the gene, develop diabetes mellitus and
skeletal defects attributed to dysfunction of insulin-
and collagen-secreting cells in the endocrine pancreas
and bone, respectively.
GENE EXPRESSION, EIF2
a
PHOSPHORYLATION AND INTEGRATION
OF
SIGNALS IN THE UPR,
AND OTHER STRESS PATHWAYS
In addition to limiting the load of client protein on the
stressed ER (by inhibiting translation of most mRNA),
eIF2
a
phosphorylation paradoxically activates the
translation of the mRNA encoding the transcription
factor ATF4 (and likely that of other regulatory
proteins which remain to be discovered, Figure 3).
PERK thus plays an important role in up-regulating
target gene expression in the UPR. Expression profiling
of ER stressed wild-type and PERK 2 /2 cells reveals a
broad role for PERK in UPR-target gene expression,
with mild impairment in most target genes and severe
impairment in a smaller group. Among the UPR target
genes that are strongly impaired in PERK 2 /2 cells are
genes involved in amino acid transport and metabolism
and a gene, GADD34, which encodes a phosphatase
that dephosphorylates eIF2
a
and terminates signaling
by PERK. The aforementioned strong PERK target
genes play an important role in maintaining translation,
by providing building blocks for secreted protein
synthesis and by promoting eIF2
a
dephosphorylation.
Thus, PERK and eIF2
a
phosphorylation appear to have
a dual role in the UPR. Early in the response they
protect the stressed cells from ER overload whereas
later they promote conditions required for synthesis of
secreted proteins.
A considerable overlap exists between genes activated
by ER stress and genes activated by other stressful
conditions that promote eIF2
a
phosphorylation.
Thus, eIF2
a
phosphorylation integrates signaling in
several stress pathways. The extent of this integrated
stress response is not fully known, but it appears to
control important aspects of cellular and organismal
metabolism.
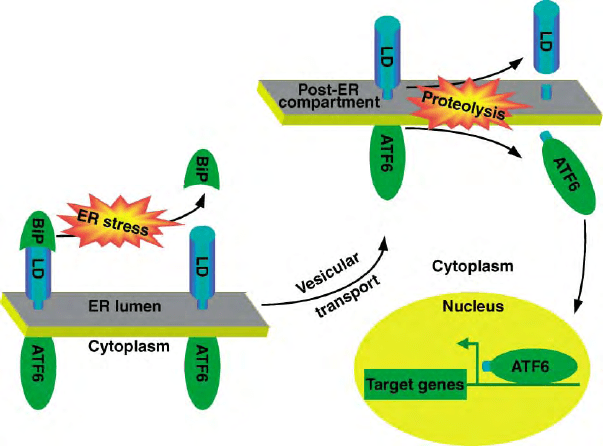
FIGURE 2 ATF6 activation by regulated intramembrane proteolysis. Membrane tethering inactivates the transcription factor ATF6, whose
effector domain is prevented from accessing its target genes in the nucleus. BiP binding to the lumenal domain of ATF6 retains the protein in the ER
compartment. However, under conditions of ER stress, BiP dissociates from ATF6, which then migrates (presumably by vesicular transport) to a
post-ER compartment that contains the proteases that liberate the effector domain to signal in the UPR.
UNFOLDED PROTEIN RESPONSES 323
A Common Mechanism
for Sensing ER Stress
The identification of three different stress sensors in the
UPR has allowed a comparison between their mode of
activation. As mentioned earlier, overexpression of BiP
and other ER chaperones had been noted to repress
signaling in the UPR. In the equilibrated ER, all three
stress transducers, IRE1, ATF6, and PERK, are found in
a simple complex with BiP, which binds on the lumenal
side to their stress-sensing domains. These complexes
are rapidly dissociated under conditions of ER stress.
Furthermore, dissociation precedes activation of the
stress transducers. BiP dissociation promotes oligomer-
ization of IRE1 and PERK, which in turns leads to
transautophosphorylation and activation of down-
stream signaling. It seems likely that BiP-dissociation
under conditions of ER stress unmasks a homo-
oligomerization domain in the similarly structured
lumenal domains of IRE1 and PERK.
The chain of events in the case of ATF6 activation is
more complex, but conceptually similar. BiP binding
retains ATF6 in the ER, segregated from the proteases
that release its transcription factor portion, as these are
localized to a post-ER compartment. BiP dissociation
liberates ATF6 to migrate to the protease-containing
compartment, a migration that presumably takes place
by vesicular transport. Thus, dispensible molecules of
BiP, which are not engaged in chaperoning ER client
proteins actively repress signaling by all three known
transducers of the UPR. It would thus appear that the
recognition of unfolded and malfolded proteins is
relegated to professional chaperones and the stress
transducers passively monitor the degree to which the
latter are engaged by their clients. This arrangement of
the upstream steps of the UPR mirrors that of the
cytoplasmic heat shock response in which unfolded
proteins in the cytoplasm activate genes implicated in
folding and degrading cytoplasmic proteins.
SEE ALSO THE FOLLOWING ARTICLES
Chaperones for Metalloproteins † Chaperones, Mole-
cular † Chaperonins † Endoplasmic Reticulum-Associ-
ated Protein Degradation † Regulated Intramembrane
Proteolysis (Rip)
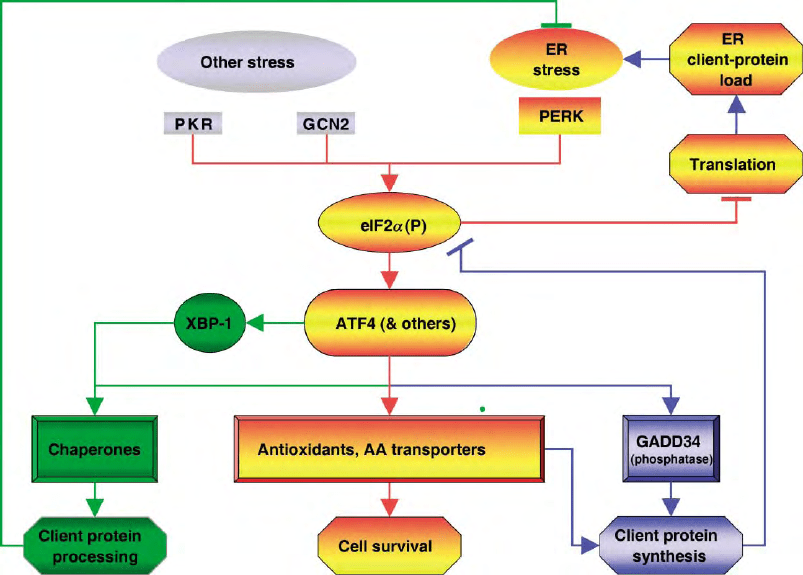
FIGURE 3 The eIF2
a
phosphorylation-dependent integrated stress response. ER stress and other stresses activate eIF2
a
kinases. Phosphorylated
eIF2 inhibits translation initiation, alleviating ER client protein load, while at the same time activating the transcription factor ATF4 (and other, yet
to be identified effectors). These activate a gene expression program with several components: The eIF2
a
phosphatase GADD34 promotes
translational recovery and resumed client protein synthesis. Genes involved in antioxidant responses and amino acid import and assimilation,
promote cell survival and client protein synthesis. PERK signaling also functions synergistically with other arms of the UPR (e.g., by transcriptional
activation of the IRE1 target gene XBP-1) to enhance ER client protein processing.
324 UNFOLDED PROTEIN RESPONSES
GLOSSARY
chaperone A protein that assists folding and assembly of other
proteins and prevents their aggregation in the unfolded state.
endoplasmic reticulum A membrane-bound organelle, present
in all nucleated cells, which serves as the entry point for
secreted proteins.
proteotoxicity A hypothetical property of unfolded and malfolded
proteins, whereby peptide domains that are illegitimately exposed
on the surface perturb cellular function.
FURTHER READING
Cox, J. S., and Walter, P. (1996). A novel mechanism for regulating
activity of a transcription factor that controls the unfolded protein
response. Cell 87, 391–404.
Harding, H., Zhang, Y., and Ron, D. (1999). Translation and protein
folding are coupled by an endoplasmic reticulum resident kinase.
Nature 397, 271–274.
Harding, H. P., Calfon, M., Urano, F., Novoa, I., and Ron, D. (2002).
Transcriptional and translational control in the mammalian
unfolded protein response. Annu.Rev.CellDev.Biol.18,
575–599.
Kaufman, R. J. (1999). Stress signaling from the lumen of the
endoplasmic reticulum: Coordination of gene transcriptional and
translational controls. Genes Dev. 13, 1211–1233.
Sidrauski, C., and Walter, P. (1997). The transmembrane kinase Ire1p
is a site-specific endonuclease that initiates mRNA splicing in the
unfolded protein response. Cell 90, 1031–1039.
Yoshida, H., Okada, T., Haze, K., Yanagi, H., Yura, T., Negishi, M.,
and Mori, K. (2000). ATF6 activated by proteolysis binds in the
presence of NF-Y (CBF) directly to the cis-acting element
responsible for the mammalian unfolded protein response. Mol.
Cell Biol. 20, 6755–6767.
BIOGRAPHY
David Ron obtained his Medical Doctorate at the Technion in Haifa,
Israel. Following residencies in internal medicine and endocrinology at
Mount Sinai Hospital and the Massachusetts General Hospital and a
research fellowship at Harvard Medical School, he took a faculty
position at New York University School of Medicine, where he is
currently a Professor of Medicine and Cell Biology. His laboratory
studies stress responses in eukaryotic cells, with a special emphasis on
signaling between organelles and the nucleus.
UNFOLDED PROTEIN RESPONSES 325

Urea Cycle, Inborn Defects of
Marsha K. Fearing and Vivian E. Shih
Harvard Medical School, Boston, Massachusetts, USA
Surplus nitrogen generated by amino acid metabolism
cannot be stored in mammals and must be excreted. The
urea cycle is the primary pathway for the disposal of
ammonium nitrogen by conversion of the toxic ammonia
molecule into innocuous urea. Inborn errors of metabolism
in the urea cycle can result in severe diseases in humans.
An accumulation of ammonia can lead to severe morbidity,
including brain damage and death.
Overview
The urea cycle is the major pathway for the disposal of
nitrogen in humans (Figure 1). Ninety percent of
ingested protein is metabolized to urea and excreted
through the urine. Ammonia is derived from a variety of
precursor protein sources. Part of the urea cycle resides
in mitochondria where ammonia is converted to
carbamoyl phosphate by carbamoyl phosphate syn-
thetase (CPS) along with its allosteric activator,
N-acetylglutamate. The next step involves carbamoyl
phosphate forming citrulline when condensed with
ornithine. This process is mediated by the enzyme
ornithine transcarbamoylase (OTC). Citrulline exits
the mitochondria and condenses with aspartate to
produce argininosuccinate. This compound is then
cleaved to arginine and fumarate by argininosuccinate
lyase. Arginine is hydrolyzed by arginase, thus releasing
urea and regenerating ornithine. Ornithine is then
shuttled into mitochondria by its own transporter.
Within the urea cycle ornithine is a conserved moiety,
it is neither formed nor lost. One atom of waste
nitrogen from ammonia and one from aspartate forms
a molecule of urea.
Liver is the only organ that contains the complete
urea cycle and is the major site of ureagenesis. Intestinal
epithelial cells have CPS and OTC for citrulline synthesis
and are the source of the circulating citrulline in plasma
that serves as a precursor for synthesis of arginine, a
semi-essential amino acid, in brain and kidney. These
organs have negligible arginase activity, hence they do
not produce urea.
Clinical Diseases
UREA CYCLE DEFECTS
Gene mutations resulting in urea cycle defects have been
described for each of the five urea cycle enzymes.
The individual disease states are N-acetylglutamate
synthetase (NAGS) deficiency, carbamoyl phosphate
synthetase (CPS) deficiency, ornithine transcarbamoy-
lase (OTC) deficiency, argininosuccinate synthetase (AS)
deficiency or citrullinemia, argininosuccinate lyase (AL)
deficiency or argininosuccinic aciduria, and arginase
(A1) deficiency.
The clinical features of the urea cycle disorders are
similar. Depending on the enzyme involved and the
residual enzyme activity, metabolic blocks in the urea
cycle will cause a resultant elevation of the plasma
ammonia level. Hyperammonemia is associated with
many of the clinical features, which include respiratory
alkalosis, seizures, acute encephalopathy, coma, and
even death. Spasticity, resembling cerebral palsy can be a
late complication. The most severe presentation is coma
in the neonatal period, but symptoms can occur later in
childhood as well. Metabolic stress, intercurrent illness,
or fever can result in endogenous protein catabolism
and precipitate a hyperammonemic crisis. In milder
cases, abnormal neuropsychiatric behavior can be the
only manifestation. Short, friable hair (trichorrhexis
nodosa) and liver fibrosis are unique to argininosucci-
nate lyase deficiency.
The exact pathophysiology of hyperammonemia is
not known and the etiology of brain edema is still
unclear. Accumulation of ammonia and astrocytic glu-
tamine are likely to be contributory to the brain damage.
Except for the X-linked ornithine transcarbamoylase
deficiency, the enzyme defects of the urea cycle disorders
are inherited as autosomal recessive traits.
The gene for OTC is located on chromosome Xp2.1.
Thus, males with OTC deficiency are usually more
severely affected with neonatal onset of hyperammone-
mia, whereas the clinical presentation in females with
OTC deficiency can be quite variable, depending on
random X-chromosome inactivation (Lyon hypothesis).
Encyclopedia of Biological Chemistry, Volume 4. q 2004, Elsevier Inc. All Rights Reserved. 326
OTHER HYPERAMMONEMIA STATES DUE
TO AMINO ACID TRANSPORTER DEFECTS
Amino acid transporter defects are disorders of com-
partmentation. The urea cycle enzymes are available
but the substrates are not transported to the proper
site, resulting in a functional enzyme deficiency.
Triple H Syndrome (Hyperornithinemia,
Hyperammonemia, Homocitrullinuria)
The HHH syndrome is a defect in ornithine mitochon-
drial transport, which prevents the recycling of
ornithine as the substrate for OTC. The end result
is a functional OTC deficiency and reduced urea
synthesis. These children present with episodic hyper-
ammonemia, ataxia, vomiting, irritability, lethargy,
and coma. They often come to attention due to
mental retardation and developmental delay, with
occasional seizure activity. The diagnostic markers
are a combination of high plasma ornithine, high
blood ammonia, increased urine orotic acid, and
high urine homocitrulline, the origin of which is
unclear. The treatment is similar to that for OTC
deficiency.
Lysinuric Protein Intolerance
Lysinuric protein intolerance (LPI), also known as
hyperdibasic aminoaciduria, is an autosomal recessive
disease with an extremely high prevalence in the Finnish
population. The gene mutation has been identified on
chromosome 14q12. LPI occurs from defects in the
cellular basolateral membrane transporter of the dibasic
amino acids, in renal tubules and the intestinal system.
This results in excessive renal loss of lysine, arginine, and
ornithine in the urine and poor gastrointestinal absorp-
tion of the same amino acids. Arginine and ornithine
deficiency impairs the function of the urea cycle and
results in hyperammonemia after protein intake. Symp-
toms of LPI include failure to thrive, vomiting, and
chronic lysine deficiency, which causes growth failure
with decreased bone density resulting in, osteoporosis.
LPI patients also have recurrent autoimmune deficiency
and infections due to low white and red blood cell
counts. Other organ involvement includes interstitial
lung disease and renal failure.
Citrin Deficiency (Citrullinemia Type II)
Citrullinemia type II was originally reported as an adult-
onset citrullinemia, mainly in Japanese patients, with
Carbamoyl
phosphate
NH
4
+
N-acetyl
glutamate
Carbamoyl phosphate
synthetase
Ornithine
transcarbamoylase
Argininosuccinate synthetase
Argininosuccinate lyase
Arginase
Glutamate
N-acetylglutamate
synthase
Citrulline
Argininosuccinate
Arginine
Ornithine
Acetyl CoA
Urea
cycle
TCA
cycle
Urea
H
2
O
Mitochondrion
Cytosol
CO
2
+
+
FIGURE 1 The urea cycle. (1) N-acetylglutamate synthetase, (2) carbamoyl phosphate synthetase, (3) ornithine transcarbamylase, (4)
argininosuccinate synthetase, (5) argininosuccinate lyase, (6) arginase, (7) mitochondrial ornithine transporter, and (8) ornithine aminotransferase.
NAG ¼ N-acetylglutamate, an allosteric activator of carbamoyl phosphate synthetase.
UREA CYCLE, INBORN DEFECTS OF 327
TABLE I
Diagnostic Characteristics of Urea Cycle Disorders and Hyperammonemic Syndromes due to Amino Acid Transporter Defects
Disorder Plasma Cit Plasma Arg Urine orotic acid
Other diagnostic
amino acids
Genetic
markers Enzyme assay Prenatal diagnosis
N-acetyl-glutamate
synthetase deficiency
# N AR; 17q21.31 Liver DNA testing
Carbamoyl phosphate
synthetase deficiency
## NAR;
chromosome 2p
Liver Linkage analysis; direct
mutational analysis
Ornithine
transcarbamoylase
deficiency
## " X linked; Xp2.1 Liver Molecular diagnosis
Argininosuccinate
synthetase deficiency
h #" AR;
chromosome 9q34
Liver; fibroblasts;
lymphoblasts,
chorionic villi
Enzyme assay; direct mutational
analysis; Metabolite
measurement in amniotic fluid
Argininosuccinate
lyase deficiency
"# ""Arginino-succinic acid
(blood and urine)
AR;
chromosome 7q
Liver; fibroblasts,
red blood cells
Metabolite measurement in
amniotic fluid
Arginase deficiency N "" AR;
chromosome 6q23
Liver, red
blood cells
Enzyme assay, DNA analysis
Lysinuric protein
intolerance
## ""arginine, lysine,
ornithine (urine)
AR;
chromosome
14q11.2
Triple H syndrome ## ""ornithine (blood)
" homocitrulline (urine)
AR;
chromosome
13q14
Fibroblasts,
Amniocytes
Enzyme assay in fetal blood,
DNA analysis
Citrin deficiency "# ""citrulline
(blood and urine)
AR;
chromosome
7q21.3
Liver DNA analysis

a liver-specific argininosuccinate synthetase deficiency.
The clinical presentation depends on the degree of
hyperammonemia and includes a sudden disturbance of
consciousness, restlessness, drowsiness, and coma with
cerebral edema. Recent studies have identified a
deficiency of the citrin protein, which is a mitochondrial
transporter of aspartate and/glutamate, as the under-
lying cause. This has been confirmed by mutation
analysis. Citrin deficiency has now been identified in
citrullinemic patients of different ethnic backgrounds
with neonatal intrahepatic cholestasis and other forms
of severe hepatic dysfunction.
Diagnosis
All disorders have hyperammonemia. Plasma and urine
amino acid analysis reveal the specific diagnostic
patterns (Table I). The amino acids proximal to the
enzyme block are increased and those distal to the block
are decreased. When there is a blockage in the urea cycle
beyond the step of CPS, the carbamoyl phosphate
synthesized cannot be used for urea synthesis and is
shunted to the biosynthetic pathway of pyrimidine to
form orotic acid. Thus, orotic acid is increased in all urea
cycle disorders except for NAGS deficiency and CPS
deficiency. Diagnosis can be confirmed by enzyme assay
in red blood cells (arginase deficiency) and cultured
fibroblasts (AS deficiency, AL deficiency). Measurement
of CPS and OTC activities can only be done in liver
biopsy. Enzymatic details can be seen in Table II.
Alternatively, molecular DNA mutation analysis can
be useful for diagnosis as well as for future genetic
counseling.
For all of the urea cycle defects, except NAGS,
prenatal diagnosis is available. AS and AL deficiencies
are noted by finding increased citrulline and increased
ASA in amniotic fluid. Amniocytes do not have all of the
urea cycle enzymes; only AS and AL are expressed. Fetal
liver biopsy is required for prenatal diagnosis of CPS and
OTC deficiencies, and fetal blood is needed for prenatal
diagnosis of arginase deficiency. For prenatal diagnosis
of HHH syndrome, the ornithine incorporation assay
can be performed in cultured amniocytes. In cases of
known mutations in a family, DNA analysis is the
preferred procedure.
Treatment
LONG-TERM MANAGEMENT
Reduction in protein intake while maintaining nutri-
tional needs to ensure growth and development remains
the mainstay of therapy. The goal of therapy is to reduce
the flow of nitrogen for disposal through the urea cycle.
This can be achieved by restriction of dietary protein
intake by providing an alternative pathway for nitrogen
disposal.
Alternative pathway therapy for nitrogen excretion
diverts ammonia clearance through compounds other
than urea. Sodium benzoate is one of the drugs used
for this purpose. When administered it conjugates with
the amino acid glycine to form hippurate, which has
high renal clearance. One mol of nitrogen is excreted
per one mole of hippurate. Following the same
principle but with greater efficiency, sodium phenyla-
cetate conjugates with the amino acid glutamine to
form phenylacetylglutamine, which eliminates two
moles of nitrogen.
To maintain normal growth it is important to provide
enough essential amino acids by supplementing the diet.
In patients with a urea cycle defect, arginine synthesis is
reduced and arginine becomes a semi-essential amino
acid. Patients can be supplemented with citrulline, the
precursor of arginine, to restore total body arginine
pools or with arginine directly in patients with AS and
AL deficiency.
In addition to medical management, liver transplan-
tation has been used with some success for treatment of
these disorders.
TABLE II
Urea Cycle Enzyme Characteristics and Locations
Enzyme Cellular location Relative activity in liver pH optimum Tissue distribution
NAGS Mitochondrial matrix 0.01 8.5 L, I, (K)
CPS1 Mitochondrial matrix 3.1 6.8–7.6 L, I, (K)
OTC Mitochondrial matrix 73 7.7 L, I, (K)
AS Cytosol 1 8.7 L, K, F, (B)
AL Cytosol 2.4 7.5 L, K, F, (B)
A1 Cytosol 962 9.5 L, RBC, (K), (B)
B ¼ brain; I ¼ small intestine; K ¼ kidney; L ¼ liver; RBC ¼ red blood cells.
UREA CYCLE, INBORN DEFECTS OF 329
ACUTE MANAGEMENT
Hyperammonemic crisis is often precipitated by stressful
situations such as fever, trauma, infection, surgery, and
dietary indiscretion. The goal of treatment is to lower
the blood ammonia levels to prevent brain edema. The
most effective treatment is hemodialysis or peritoneal
dialysis. Nitrogen scavenger medications, such as
sodium phenylacetate and sodium benzoate, and argi-
nine are also given intravenously. Protein intake is halted
and an intravenous fluid with a high dextrose (10%)
load is provided immediately to minimize catabolism
and therefore the source of ammonia. Intercurrent
infection should be treated appropriately.
Future Horizons
Future treatment modalities may include total or partial
organ transplant, stem cell transplant, and enzyme
replacement. Early diagnosis utilizing newborn screen-
ing and prenatal diagnosis will allow early treatment
and improve the outcome.
SEE ALSO THE FOLLOWING ARTICLES
Amino Acid Metabolism † Ornithine Cycle
GLOSSARY
autosomal recessive A mode of genetic inheritance that describes a
trait or disorder requiring the presence of two copies of a gene
mutation on one of the 22 pairs of autosomes (nonsex chromo-
somes) at a particular locus in order to express observable
phenotype.
enzyme Biological reactants that are protein catalysts, mediating
reactions without themselves being changed in the overall process.
They selectively channel substrates into pathways.
essential amino acids The amino acids that are synthesized by
nonmammalian pathways and must be obtained in diet. They
include histidine, isoleucine, leucine, lysine, methionine, phenyl-
alanine, threonine, tryptophan, and valine. Arginine is considered a
“semi-essential” amino acid because mammals synthesize arginine,
but cleave most of it to form urea and require greater amounts
than can be produced in mammalian biosynthesis.
Lyon hypothesis The X chromosome is randomly inactivated in early
development in embryonic cells. This results in fixed inactivation in
the female’s descendant cells. The deactivated chromosome forms
the Barr body.
non-essential amino acids Amino acids that can be produced by
mammals from common intermediates and biosynthesis and are
not strictly diet dependent. These include alanine, asparagine,
aspartate, cysteine, glutamate, glutamine, glycine, proline, serine,
and tyrosine.
X-linked disorder A mode of genetic inheritance that describes a gene
mutation on the X chromosome that causes the phenotype to be
expressed in males who are hemizygous for the gene mutation and
in females who are homozygous for the gene mutation (meaning
they must have a copy of the gene mutation on each of their two
X chromosomes). Carrier females do not usually express the
phenotype, although differences in X-chromosome inactivation can
lead to varying degrees of clinical expression.
FURTHER READING
Berry, G., and Steiner, R. (2001). Long-term management of patients
with urea cycle disorders. J. Pediatrics 138(Suppl. 1), S56–S61.
Feillet, F., and Leonard, J. V. (1998). Alternative pathway therapy for
urea cycle disorders. J. Inher. Metab. Dis. 21(Suppl. 1), 101 –111.
Fernandes, J., Saudubray, J. M., and Van den Berghe, G. (eds.) (2000).
Inborn Metabolic Diseases: Diagnosis and Treatment, 3rd edition.
Springer, Berlin.
Morizono, H., Caldovic, L., Shi, D., and Tuchmon, D. (2004).
Mammalian N-acetylglutamate synthase. Mol. Genet. Metab. 81,
4–11.
Nyhan, W. L., and Ozand, P. T. (eds.) (1998). Atlas of Metabolic
Diseases. Chapman and Hall Medical, London.
Steiner, R., and Cederbaum, S. (2001). Laboratory evaluation of urea
cycle disorders. J. Pediatrics 138(Suppl. 1), S21–S29.
Summar, M., and Tuchman, M. (2001). Proceedings of a consensus
conference for the management of patients with urea cycle
disorders. J. Pediatrics 138(Suppl. 1), S6–S10.
BIOGRAPHY
Marsha K. Fearing is a Clinical Fellow at the Harvard Medical School
Genetics Training Program, specializing in Biochemical Genetics. She is
a member of the Scholars in Clinical Science Program at Harvard
Medical School. Her area of research is translational and public health
research in metabolism. Dr. Fearing is a member of Alpha Omega
Alpha, the American Academy of Pediatrics, and a candidate fellow of
the American College of Medical Genetics.
Vivian E. Shih is Professor of Neurology at Harvard Medical School,
Associate Neurologist and Pediatrician at Massachusetts General
Hospital, and Director of the Clinical Neurochemistry and Amino Acid
Disorders Laboratory at Massachusetts General Hospital. Her area of
research is inborn errors of amino acid and organic acid metabolism.
330 UREA CYCLE, INBORN DEFECTS OF

Vacuoles
Christopher J. Stefan and Scott D. Emr
University of California, San Diego, La Jolla, California, USA
Vacuoles and their mammalian counterparts, lysosomes, are
membrane-bound cytoplasmic organelles that contain an
assortment of soluble acid-dependent hydrolases and a set of
highly glycosylated integral membrane proteins. Most notably,
this organelle is an important site for the degradation of
cellular lipids, membrane-associated proteins, and cytoplasmic
proteins. In addition to its degradative functions, the vacuo-
le/lysosome plays an important role in pH and ion homeostasis
and is a site for the generation/storage of nutrients, amino
acids, antigens, and additional signaling factors.
Identification/Discovery of
Vacuoles and Vacuolar Constituents
Vacuoles/lysosomes were first visualized by independent
cell morphological studies undertaken in the 1950s by
the Palade and Novikoff laboratories. These studies
revealed that lysosomes appear as organelles with
0.5 mm diameter and a heterogeneous morphology,
containing electron-dense cores and, in some cases,
membrane vesicles. In the yeast Saccharomyces cerevi-
siae, vacuoles are larger with respect to cell size, but
display similar characteristics in terms of electron
density and intralumenal vesicles, when observed
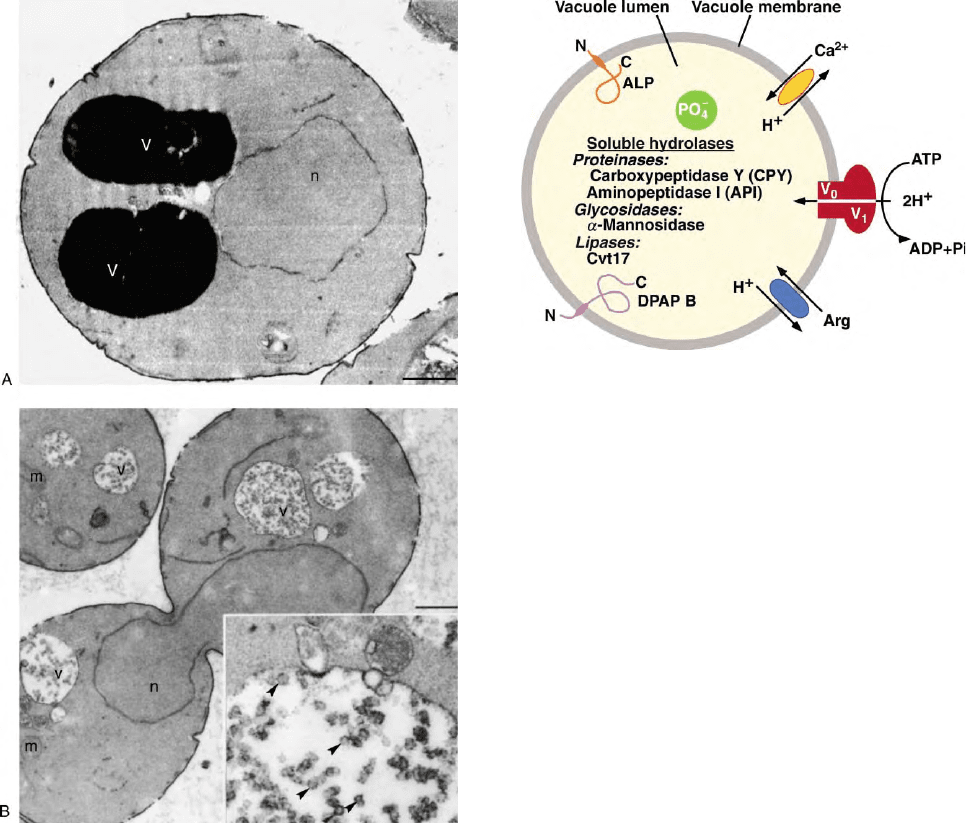
under certain conditions (Figures 1A and 1B).
Biochemical studies performed by de Duve and
colleagues initially identified lysosomes as organelles of
highly buoyant centrifugal properties that contained a
number of hydrolytic activities. The hydrolases within
this fraction demonstrated attenuated activity depen-
dent on the integrity of their surrounding membrane.
Specifically, hydrolase activity was retained within the
limiting vacuole/lysosome membrane, restricted from
the cell lysate unless the limiting membrane was
disrupted. Since vacuoles/lysosomes are morphologi-
cally heterogeneous, biochemical studies have produced
the current definition of vacuoles/lysosomes as mem-
brane-bound acidic organelles that contain mature
acid-dependent hydrolases and certain integral mem-
brane-glycoproteins. Vacuoles lack other proteins that
distinguish them from endosomes (compartments that
deliver material to vacuoles), such as biosynthetic
sorting receptors and recycling cell-surface receptors.
As already mentioned, vacuoles/lysosomes are mem-
brane compartments that serve as a major degradative
compartment in eukaryotic cells. Both endogenous
and exogenous molecules can be delivered to vacuoles
through biosynthetic and endocytic pathways. In addi-
tion, vacuoles can degrade proteins transported from the
cytosol. The degradative function of these organelles
is carried out by numerous acid-dependent hydrolases
(e.g., proteases, lipases, glycosidases) contained within
its lumen (Figure 2). The outer, limiting membrane of
vacuoles also contains a set of highly glycosylated,
vacuolar/lysosomal-associated membrane proteins.
A subset exhibits known hydrolase activities (Figure 2),
while the functions of many are still unclear. Additional
vacuolar membrane proteins mediate transport of ions,
amino acids, and other solutes across the vacuolar
membrane and maintain an acidic lumenal pH in the
range of 4.5–6.5 (Figure 2).
Vacuole Function in Yeast
The vacuole of the yeast S. cerevisiae plays an important
role in protein degradation, pH regulation, ion homeo-
stasis, and nutrient storage. The yeast vacuole contains a
large number of hydrolases (proteases, lipases, glycosi-
dases; see Figure 2) with various substrate specificities.
Thus, it is a major site for degradation of cellular
proteins, lipids, and even whole organelles.
A remarkable feature of vacuoles is their acidifica-
tion. In yeast, a specific vacuolar proton pump ATPase
(V-ATPase) mediates this process. Biochemical and
genetic approaches have identified 14 subunits of the
V-ATPase (encoded by the VMA genes for vacuolar
membrane ATPase). The V-ATPase subunit proteins
form two complexes, the peripheral V
1
subcomplex,
responsible for ATP hydrolysis, and the V
0
subcomplex,
responsible for proton translocation across the mem-
brane (Figure 2). Cells lacking a functional V-ATPase are
viable, but require an intact endocytic pathway to the
vacuole. Moreover, they fail to grow on media with
neutral pH, indicating that vacuole acidification is an
v
Encyclopedia of Biological Chemistry, Volume 4. q 2004, Elsevier Inc. All Rights Reserved. 331
essential feature. Consistent with this, the electrochemi-
cal gradient generated by the V-ATPase is necessary to
drive several other transport systems described in this
article (Figure 2).
In addition to its hydrolytic functions, the vacuole
serves as a storage compartment for several ions and
nutrients (Figure 2). For example, the Vcx1 protein acts
as a high capacity H
þ
/Ca
2þ
exchanger in vitro and has
been implicated in the regulation of vacuolar calcium
flux in vivo (Figure 2). The vacuole also stores both
phosphate and polyphosphate (Figure 2). Since hydroly-
sis of phosphate leads to the release of protons, a role for
polyphosphate in pH homeostasis has been suggested.
Polyphosphate is proposed to function as a cation
counter ion as well. Some amino acids are also stored
inside the vacuole at high concentrations. For example,
arginine accumulates in vacuoles at concentrations
nearly an order of magnitude greater than in the
surrounding media (Figure 2). Consistent with this,
several H
þ
/amino acid anti-porter systems have been
identified in yeast. Pools of amino acids stored in the
vacuole may be utilized during nitrogen starvation.
Vacuole Biogenesis and Transport
Pathways to the Vacuole in Yeast
To carry out the numerous functions that vacuoles
perform, several transport pathways have evolved that
deliver cellular components to vacuoles. Model genetic
systems such as the yeast S. cerevisiae have been
instrumental in identifying both proteins and lipids
that constitute components of vacuoles and the machin-
ery involved in transport to vacuoles. Importantly, many
of these factors and transport pathways are conserved in
mammalian cells.
At least eight pathways have been identified in
vacuole biogenesis and protein transport to vacuoles in
FIGURE 2 Schematic overview of vacuolar components. The
vacuole lumen possesses several hydrolase activities and accumulates
ions, such as phosphate (PO
4
2
) and polyphosphates. The vacuolar
membrane also contains known hydrolases, such as ALP, and
proteases, such as dipeptidyl-aminopeptidase B (DPAP B). The
vacuolar ATPase (V
0
/V
1
) functions as the primary proton pump that
generates an electrochemical gradient used to drive other vacuole
membrane transport systems, such as arginine (Arg) and divalent
cation (Ca
2þ
) anti-transporters.
FIGURE 1 Ultrastructural analysis of vacuole morphology in wild
type (A) and hydrolase deficient (B) yeast cells reveals electron dense
cores and intralumenal membrane vesicles, respectively. (A) Wild type
cells were grown to mid-log phase, fixed, and visualized by electron
microscopy. Vacuoles appear as dark, electron dense organelles due
to the accumulation of cellular proteins, lipids, and other molecules.
(B) In cells lacking vacuolar ATPase activity (vma4D mutant cells),
vacuoles appear translucent by electron microscopy and contain
intralumenal vesicles. Inset: High magnification image of vacuolar
vesicles (arrowheads) (v, vacuoles; n, nucleus; m, mitochondria;
bars ¼ 0.1 mm). (Reprinted with permission from Wurmser, A. E.,
and Emr, S. D. (1998). Phosphoinositide signaling and turnover:
PtdIns(3)P, a regulator of membrane traffic, is transported to the
vacuole and degraded by a process that requires lumenal vacuolar
hydrolase activities. The EMBO Journal 17, 4930– 4942, copyright
1998 Oxford University Press.)
332 VACUOLES