Lennarz W.J., Lane M.D. (eds.) Encyclopedia of Biological Chemistry. Four-Volume Set . V. 4
Подождите немного. Документ загружается.

for a short-term treatment. Recent experimental evi-
dence strongly suggests that such inhibitors may indeed
be beneficial in certain pathologies, such as in multiple
myeloma, a malignancy that affects the bone marrow.
A completely different approach to drug development
can be the development of small molecules that interfere
with the activity of specific E3s. For example, peptides
that bind specifically to HPV-E6 can prevent its
association with p53 and interfere with its targeting by
E6-AP. Such peptides were able to induce p53 in HPV-
transformed cells with subsequent reversal of certain
malignant characteristics and induction of apoptosis.
SEE ALSO THE FOLLOWING ARTICLES
26S Proteasome, Structure and Function † Ornithine
Cycle † Proteasomes, Overview † Protein Degradation †
SUMO Modification † Ubiquitin-Like Proteins
GLOSSARY
proteolysis/degradation Hydrolysis of a protein which is a hetero-
polymer of amino acids to short peptides, which contain a few
amino acids, and/or to single amino acids.
ubiquitination Covalent modification of a protein by the small
protein ubiquitin.
FURTHER READING
Glickman, M. H., and Ciechanover, A. (2002). The ubiquitin –
proteasome proteolytic pathway: Destruction for the sake of
construction. Physiol. Rev. 82, 373–428.
Goldberg, A. L., Elledge, S. J., and Wade, J. (2001). The cellular
chamber of doom. Sci. Am. January, 68–73.
Hilt, W., and Wolf, D. H. (eds) (2000). Proteasomes: The World of
Regulatory Proteolysis. Eurekah.com/LANDES BIOSCIENCE
Publishing Company, Georgetown, Texas, USA.
Weissman, A. M. (2001). Themes and variations on ubiquitylation.
Nat. Rev. Cell Mol. Biol. 2, 169–179.
BIOGRAPHY
Aaron Ciechanover is a Distinguished Professor in the Cancer and
Vascular Research Center in the Faculty of Medicine at the
Technion, Haifa, Israel. His principal research interests are
regulation of transcriptional factors by the ubiquitin system.
He holds an M.D. from “Hadassah” Medical School of the
Hebrew University in Jerusalem, and a Ph.D. in biochemistry from
the Technion. While a graduate student of Dr. Avram Hershko, the
two discovered the principles and machinery of ubiquitination as a
degradation signal.
Michael Glickman is an Associate Professor in the Department of
Biology at the Technion. His principal research interest is mechanisms
of proteolysis by the proteasome, and proteasome composition.
He holds a Ph.D. in chemistry from the University of California at
Berkeley. As a research fellow with Dr. Dan Finley at Harvard Medical
School, he uncovered the basic structure of the regulatory complex of
the proteasome.
UBIQUITIN SYSTEM 303

Ubiquitin-Like Proteins
Edward T. H. Yeh
The University of Texas, Houston, Texas, USA
Ubiquitin is a small polypeptide that covalently attaches to
other proteins, which marks them for degradation by the
proteasome. Ubiquitin regulates many important cellular
processes, such as signal transduction, cell-cycle progression,
and the transformation of normal to malignant cells. Several
ubiquitin-like proteins, such as sentrin (also called SUMO)
and NEDD8/Rub1 have also been discovered. Similar to
ubiquitin, these ubiquitin-like proteins also modify other
proteins. However, they do not trigger proteasomal degra-
dation. Sentrin, in a process called sentrinization, usually
produces a change of the target protein’s cellular localization
and function. NEDD8, with cullin as its substrate, is a key
component of the ubiquitin ligase complex. Thus, NEDD8,
in a process called neddylation, indirectly regulates the
ubiquitin/proteasome system. Sentrinization and neddylation
require unique enzymes – E1, E2, and E3 – that are distinct
from those involved in the ubiquitin pathway. Furthermore,
both sentrinization and neddylation can be regulated by
proteases that specifically remove sentrin or NEDD8 from
their cellular targets.
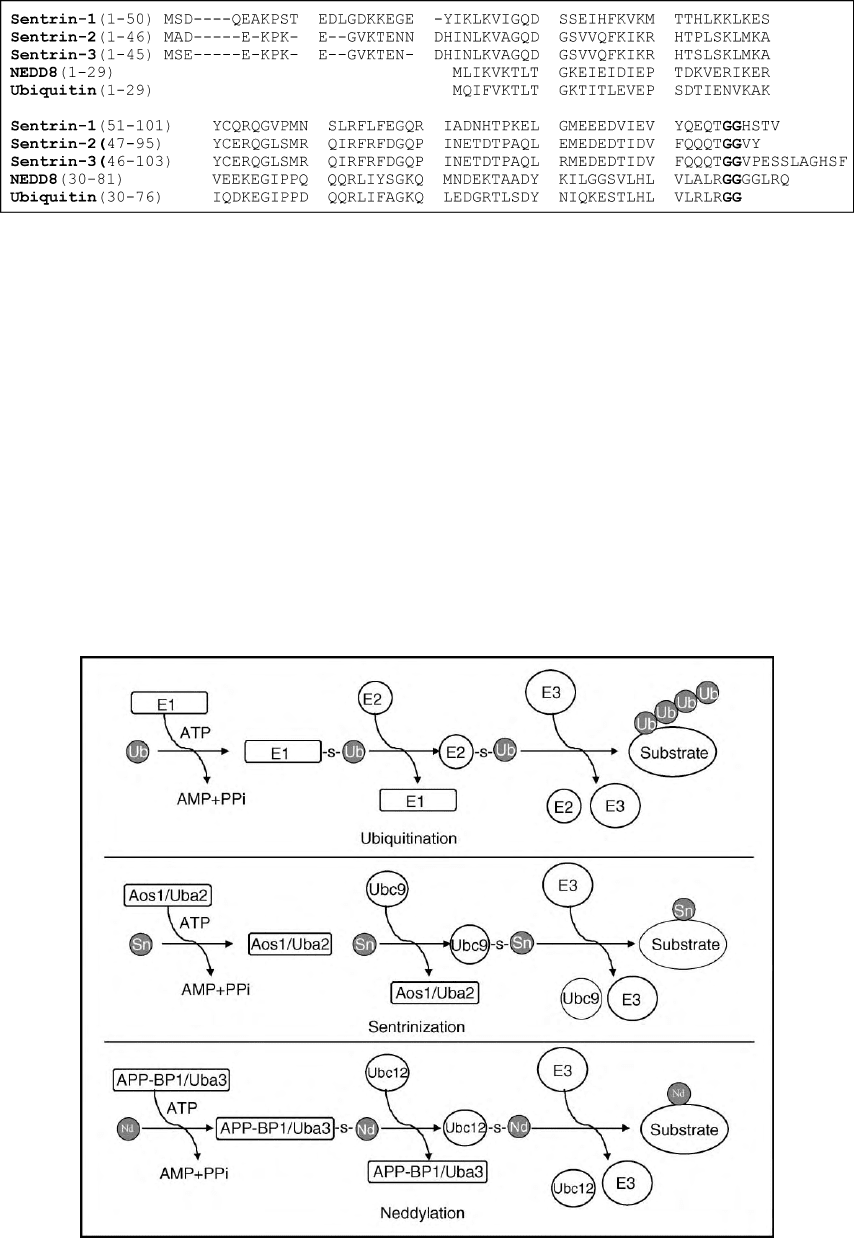
Sentrin
There are three sentrins. Sentrin-1 is a 101-amino-acid
protein containing a ubiquitin homology domain
(residues 22–97) that is 48% homologous to human
ubiquitin (see Figure 1). Sentrin-2 is a 95-amino-acid
polypeptide that is 66% similar to sentrin-1 in its
ubiquitin homology domain. Sentrin-3 is a 103-amino-
acid polypeptide that is 97% identical to sentrin-2 in its
ubiquitin homology domain. All sentrins have distinct
N-terminal amino-acid sequences and C-terminal exten-
sions. In addition, the Gly–Gly residues required for
sentrinization are conserved in all sentrins (Figure 1).
ACTIVATION, CONJUGATION,
AND
LIGATION OF SENTRINS
The amino acids following the conserved Gly–Gly
residues in all sentrins are cleaved by C-terminal
hydrolases. The resultant sentrin monomer is then
transferred to a specific E1 complex (Figure 2). The
activating enzyme (E1) for sentrin is composed of two
subunits, AOS1 and UBA2. The human AOS1 protein is
56% similar to the N-terminal half of the ubiquitin E1.
The human UBA2 protein, which is homologous to the
C-terminal half of the ubiquitin E1, contains the active-
site cysteine residue required for the formation of a thiol
ester linkage with sentrins. The activated sentrin
molecule attached to E1 is then transferred to a carrier
protein, E2.
In contrast to the large number of E2s involved in
ubiquitination, sentrin utilizes only UBC9 as the carrier
protein. This exclusive usage of UBC9 is probably
explained by the existence of a 5-residue insertion that
forms an exposed tight
b
-hairpin and a 2-residue
insertion that forms a bulge in a loop close to the active
site of UBC9. The surface of UBC9 involved in sentrin-1
binding is positively charged, whereas the corresponding
surface in sentrin-1 is negatively charged. Moreover, the
face of UBC9 remote from the catalytic site possesses a
distinct electrostatic potential that may account for the
propensity of UBC9 to interact with other proteins.
There are at least three different E3 involved in
sentrinization. RanBP2, a protein that has been localized
in the nuclear pore complex, serves as the E3 for Sp100
and histone deacetylase 4. RanBP2 also binds to UBC9,
suggesting that sentrinization could take place at the
nuclear pore complex. Another E3, PIAS (protein
inhibitor of activated STAT), promotes sentrinization
of the androgen receptor, c-JUN, p53, and Sp3. Finally,
the human polycomb group protein, Pc2, functions as an
E3 for a generalized transcription repressor, CtBP.
PROTEINS MODIFIED BY SENTRIN
The list of sentrinized proteins has been expanding
rapidly. Three examples that have been characterized are
discussed here, viz., PML, RanGAP1, and I
k
B
a
.
PML, a RING finger protein with tumor suppressor
activity, has been implicated in the pathogenesis of acute
promyelocytic leukemia, which arises following a
reciprocal chromosomal translocation that fuses the
PML gene with the retinoic acid receptor
a
(RAR
a
)
gene. PML can be modified by all sentrins, but not by
ubiquitin or NEDD8. Two forms of PML-RAR
a
fusion
proteins have been found to be expressed in acute
Encyclopedia of Biological Chemistry, Volume 4. q 2004, Elsevier Inc. All Rights Reserved. 304
promyelocytic leukemia. Remarkably, neither can be
sentrinized in vivo. Extensive mutational analysis of
PML has shown that Lys65 in the RING finger domain,
Lys160 in the B1 Box, and Lys490 in the nuclear
localization signal constitute three major sentrinization
sites. The PML mutant with Lys to Arg substitutions in
all three sites is expressed normally but cannot be
sentrinized. It also cannot recruit Daxx to the nuclear
body. The lack of sentrinization of PML eliminates
PML’s transcriptional coactivator activity.
RanGAP1, a major regulator of the Ras-like GTPase
Ran, which plays a critical role in the bidirectional
transport of proteins and ribonucleoproteins across the
nuclear pore complex was the first protein shown to be
modified by sentrin-1. The 90 kDa sentrinized form of
RanGAP1 associates with RanBP2 of the nuclear pore
complex, whereas the 70 kDa unmodified form of
RanGAP1 is exclusively cytoplasmic. Thus, sentriniza-
tion is critical for RanGAP1 to translocate to the nuclear
pore. Interestingly, RanGAP1 cannot be modified by
sentrin-2/3.
Although most sentrinized proteins are localized in the
nucleus or associated with the nuclear envelope, a cyto-
solic protein, I
k
B
a
, can also be modified by sentrin-1.
FIGURE 1 Alignment of sentrin-1, sentrin-2, sentrin-3, NEDD8, and ubiquitin. The conserved C-terminal glycine–glycine residues were printed
in bold.
FIGURE 2 Schematic diagram of ubiquitination, sentrinization, and neddylation. E1: activating enzyme; E2: conjugating enzyme; E3: ligases.
The E1 for the sentrinization pathway consists of Aos1 and Uba2, whereas the E1 for the neddylation pathway consists of APP-BP1 and Uba3. The
E2 for the sentrinization pathway is Ubc9, whereas the E2 for the neddylation pathway is Ubc12. There are multiple E2s for the ubiquitination
pathway. Ub: ubiquitin; Sn: sentrin: Nd: NEDD8.
UBIQUITIN-LIKE PROTEINS 305
I
k
B
a
is a cytosolic inhibitor of NF
k
B, a transcription
factor involved in the induction of a large number of
proteins involved in inflammation. I
k
B
a
is phosphory-
lated on serine residues 32 and 36 following tumor
necrosis factor stimulation. The phosphorylated form of
I
k
B
a
is then polyubiquitinated on lysine residues 21 and
22 and degraded by the proteasome. Unlike ubiquitina-
tion, sentrinization of I
k
B
a
on lysine residue 21 is
inhibited by prior phosphorylation. Furthermore, sen-
trinized I
k
B
a
cannot be ubiquitinated and is resistant to
proteasomal degradation. Thus, sentrinization can com-
pete with ubiquitination for the same Lys residue on
target proteins, effectively constitutes an antiubiquitin
process.
By comparing the amino-acid sequences surrounding
the acceptor lysine residues, a sentrinization consensus
sequence can be formulated. It appears that the acceptor
Lys residue is preceded by a hydrophobic amino acid (Ile
or Leu) and followed by a polar amino acid, such as Gln,
Thr, and a charged amino acid, Glu. The general
consensus formula is (I/L)K(Q/T)E.
DE-SENTRINIZATION
In yeast, there are two sentrin-specific proteases (SENPs)
that deconjugate sentrin from its substrate. Ulp1
possesses both isopeptidase and C-terminal hydrolase
activity and is essential for the transition from G2 to the
M phase of the cell cycle, the same phenotype observed
for the Ubc9 mutation in yeast. These observations
suggest that sentrinization and de-sentrinization are
required for cell-cycle progression in yeast. Ulp2, on
the other hand, is not essential for cell viability. The
Ulp2 mutant exhibits a pleiotropic phenotype that
includes temperature-sensitive growth, abnormal cell
morphology, and decreased plasmid and chromosome
stability. The mutant is also hypersensitive to DNA-
damaging agents, such as hydroxyurea. Ulp1 is localized
in the nuclear envelope, whereas Ulp2 is in the nucleus.
In humans, there are three well-characterized SENPs.
All human SENPs have conserved C-terminal region,
even though they vary in size. The similarity between
yeast Ulp1, Ulp2, and human SENPs is confined
primarily to the C-terminal region of , 200 amino
acids, within which a ,90-residue segment forms a core
structure common to cysteine proteases. It appears that
the conserved C termini of the SENPs contain the
catalytic domain, whereas the N termini regulate their
cellular localization and substrate specificity. SENP1 is
localized in the nucleus, excluding the nucleolus; SENP2
is localized in the nuclear envelope and also forms
nuclear speckles; and SENP3 is localized predominantly
in the nucleolus. Both SENP1 and SENP2 can deconju-
gate sentrinized PML but not RanGAP1. In addition,
SENP1 and SENP2 can deconjugate many sentrinized
nuclear proteins. However, SENP3 appears to have more
restricted substrate specificity.
In summary, the sentrin system possesses its own E1,
E2, E3, and deconjugating enzymes, which serve to
regulate an increasingly recognized cellular processes.
NEDD8/Rub1
NEDD8 is a small protein consisting of 81 amino
acids that is 80% homologous to ubiquitin (see
Figure 1). The yeast and plant homologues of
NEDD8 have been termed Rub1 (related to ubiquitin
1) and are processed and conjugated to a small
number of cellular proteins. The overall structures of
NEDD8 and Rub1 are quite similar to those of
ubiquitin. The major difference occurs in two surface
regions with a different electrostatic surface potential.
Thus, unlike sentrin-1, NEDD8 is structurally more
closely related to ubiquitin.
ACTIVATION AND CONJUGATION
OF
NEDD8 AND RUB1
UCH-L3, one of the ubiquitin C-terminal hydrolases,
also binds to NEDD8 and functions as a NEDD8
C-terminal hydrolase. Following processing of its C
terminus, NEDD8 is transferred to its E1.
The E1 for human NEDD8 is composed of two
subunits, APP-BP1 and UBA3. The human APP-BP1
protein is 56% similar to the N-terminal half of
ubiquitin’s E1. The human UBA3 protein, which is
homologous to the C-terminal half of ubiquitin’s E1,
contains the active-site cysteine residue required for the
formation of thiol ester linkage with NEDD8. A similar
E1 has also been identified in yeast and plants. The
counterpart of APP-BP1 has been named AXR1 in plant.
The importance of the Rub1 conjugation system is
highlighted by the requirement of the AXR1 protein for
the normal response to the plant hormone auxin in
Arabidopsis thaliana. The specificity of NEDD8’s E1 is
due, in part, to the preferential binding of UBA3 to
NEDD8, but not to ubiquitin or sentrin-1. The
conjugating enzyme for NEDD8 or Rub1 is UBC12. A
dominant-negative form of UBC12 could abolish
NEDD-8 conjugation in vivo and inhibit cell growth.
RUB1/NEDD8 MODIFICATION OF
CDC53/CULLINS
The major substrate for Rub1 in yeast is Cdc53 (also
known as yeast cullin), a 94 kDa protein required for the
G
1
to S phase of the cell-cycle progression. Cdc53 is a
common subunit of the SCF complex, a ubiquitin ligase
(E3) composed of Skp1, Cdc53/cullin, and an F box
306
UBIQUITIN-LIKE PROTEINS
protein. There are many F-box proteins, which play a
critical role in conferring substrate specificity to the
SCF complex. For example, the F-box protein, Cdc4,
forms a SCF
Cdc4
complex with Skip1 and Cdc53/cullin
that binds to phosphorylated Sic1 and catalyzes its
ubiquitination.
In mammalian cells, NEDD8 modifies a limited
number of human cellular proteins that primarily
localized in the nucleus. Interestingly, all of the known
NEDD8 targets in mammalian cells are cullins. Human
Cul-1 is a major component of the SCF complex that is
involved in the degradation of I
k
B
a
,
b
-catenin, and p27.
The neddylation of Cul-1 dissociates CAND1, an
inhibitor of Cul-1–SKP1 complex formation. The
suppression of CAND1 therefore increases the level of
the CuL-1–SKP1 complex. Thus, the neddylation of
Cul-1 enhances the formation of the SCF complex,
which in turn stimulates protein polyubiquitination.
Human Cul-2 binds to the von Hippel-Lindau gene
product through elongin B and elongin C to form a
complex (Cul-2–VBC complex) that appears to have
ubiquitin ligase activity. The VBC complex itself
promotes, but is not essential for, NEDD8 conjugation
to Cullin-2. Human Cul-3 was initially identified as a
salicylate suppressible protein with unknown mechan-
ism of activation, and was recently shown to be involved
in the ubiquitination of cyclin E to control the S phase in
mammalian cells. Human Cul-4A associates with UV-
damaged DNA-binding protein and may play a role in
DNA repair. Thus, the cullins may constitute a family of
ubiquitin E3 ligases that have diverse biologic functions,
with the neddylation of cullins playing a crucial role in
ligase activity.
REGULATION OF NEDD8-CONJUGATES
There are several proteins that regulate NEDD8 con-
jugates. USP21 is a ubiquitin-deconjugating enzyme
that is also capable of removing NEDD8 from NEDD8
conjugates. The overexpression of USP21 profoundly
inhibits growth of U2OS cells. DEN1 is a specific protease
for NEDD8 conjugates, as it has no activity against
ubiquitin or sentrin conjugates. DEN1 can de-neddylate
NEDD8 conjugated Cul-1. Interestingly, DEN1 is highly
homologous to the catalytic domain of SENP. The
COP9 signalosome (CSN) is an evolutionarily conserved
multiprotein complex composed of eight subunits.
It was first identified as an essential component in
the repression of light-regulated development in
Arabidopsis. CSN interacts with the SCF-type E3
ubiquitin ligases containing Cul1, which suggests that
CSN and SCF-type E3 ubiquitin ligases are closely
related. Indeed, CSN promotes the cleavage of NEDD8
from neddylated Cul1. The Jab1/MPN domain metal-
loenzyme motif in Csn5 subunit is responsible for the
de-neddylation activity of CSN. Thus, CSN binds to SCF-
type E3 ubiquitin ligases and probably regulates their
activity through the de-neddylation of conjugated Cul-1.
NUB1, an interferon-inducible protein, also regulates the
neddylation system. The overexpression of NUB1 leads
to the disappearance of most neddylated proteins. NUB1
appears to act by recruiting NEDD8 and its conjugates to
the proteasome for degradation, further highlighting the
close functional tie between neddylation and
ubiquitination.
SEE ALSO THE FOLLOWING ARTICLES
Proteasomes, Overview † SUMO Modification †
Ubiquitin System
GLOSSARY
neddylation The process of modifying a substrate with NEDD8.
proteasome A cellular organelle specialized in degrading ubiquitin-
conjugated proteins.
sentrinization The process of modifying a substrate with
sentrin/SUMO.
ubiquitination The process of modifying a substrate with ubiquitin.
FURTHER READING
Hay, R. T. (2001). Protein modification by SUMO. Trends Biochem.
Sci. 26, 332 –333.
Melchior, F. (2000). SUMO – nonclassical ubiquitin. Annu. Rev. Cell
Dev. Biol. 16, 591–626.
Yeh, E. T. H., Gong, L., and Kamitani, T. (2000). Ubiquitin-like
proteins: New wines in new bottles. Gene 248, 1–14.
BIOGRAPHY
Edward T.H. Yeh is Chairman of the Department of Cardiology at the
University of Texas M.D. Anderson Cancer Center and Director of the
Research Center for Cardiovascular Diseases at the Institute of
Molecular Medicine at the University of Texas-Houston Health
Science Center. He holds an M.D. from the University of California,
Davis and received his postdoctoral training at Harvard University. His
laboratory was among the first to show that sentrin and NEDD8 can be
covalently conjugated to other proteins.
UBIQUITIN-LIKE PROTEINS 307

UmuC, D Lesion Bypass
DNA Polymerase V
Zvi Livneh
Weizmann Institute of Science, Rehovot, Israel
DNA polymerase V from the bacterium Escherichia coli
(E. coli) is specialized to perform DNA synthesis across DNA
lesions. The latter are sites in DNA that were chemically
damaged by radiation or chemicals, and which usually block
DNA replication. The reaction catalyzed by pol V is called
translesion synthesis (TLS), translesion replication (TLR), or
lesion bypass. It is usually mutagenic, since the coding
properties of damaged nucleotides in DNA are often different
from those of the original undamaged nucleotides. Pol V is a
heterotrimer composed of one molecule of the UmuC protein
(48 kDa), and two molecules of the UmuD
0
protein (12 kDa
each). UmuD
0
is a proteolytic cleavage product of a larger
protein, termed UmuD (15 kDa). The UmuD and UmuC
proteins are encoded by an operon that is induced by DNA-
damaging agents, under regulation of the SOS response. Pol V
is a low-fidelity DNA polymerase, which does not have an
exonuclease proofreading activity, and exhibits low processiv-
ity. Its lesion-bypass activity requires two additional proteins,
RecA, which is also the main recombinase in E. coli, and
single-strand DNA-binding protein (SSB), a protein essential
for replication. In addition, pol V is stimulated by the
processivity proteins of pol III, namely, the
b
-subunit DNA
sliding clamp, and the
g
-complex clamp loader. By virtue of its
mutagenic lesion-bypass activity, pol V is responsible for the
formation of mutations by a variety of DNA-damaging agents
that cause replication blocks, such as UV radiation, methy-
methanesulphonate (MMS), and more. Pol V homologues
were conserved in evolution from E. coli to humans, and
comprise the Y family of DNA polymerases.
Pol V and the SOS Response
The SOS response is a global stress response in E. coli,
induced by DNA damage that causes interruption of
DNA replication. It involves the induction of 30–40
genes, which collectively act to increase cell survival
under adverse environmental conditions. SOS genes
comprise a regulon, whose members are negatively
regulated at the transcriptional level by the LexA res-
pressor. Induction involves autocleavage of free LexA
repressor, promoted by an activated form of the RecA.
This cleavage shifts the binding equilibrium of LexA,
causing it to dissociate from its target genes, thereby
causing induction. RecA is usually activated by forming
a complex of multiple RecA molecules bound to a
stretch of single-stranded DNA (ssDNA), which is
formed as a result of exposure to damaging agents.
This RecA nucleoprotein filament has multiple roles in
cellular responses to DNA damage. In addition to the
induction of the SOS regulon, it promotes homologous
recombination repair, and it is essential for lesion bypass
by pol V. The umuD and umuC genes, encoding the two
subunits of pol V, are arranged in an operon that is part
of the SOS regulon (namely, its transcription is regulated
by LexA and RecA), and are therefore inducible by
DNA-damaging agents such as UV radiation or MMS.
Mutations that inactivate umuC or umuD cause a
drastic decrease in UV- or MMS-mutagenesis, with a
minor effect on cell survival. This demonstrated the
surprising phenomenon that mutagenesis caused by
DNA-damaging agents is an inducible process, which
requires specific gene products. In other words, muta-
genesis is not merely a by-product of replication, and the
presence of lesions in DNA does not cause mutations
without the activity of specific inducible proteins.
The Discovery of Pol V
It took nearly two decades from the time the umuC and
umuD genes were cloned, until the discovery that they
encode a specialized lesion-bypass DNA polymerase.
This long period of time is attributed to the great
difficulty in obtaining UmuC in soluble and active form,
the lack of any amino acid homology to classical DNA
polymerases, and the complexity of the in vitro lesion-
bypass assay system. In addition, genetic experiments
had indicated that the polC (dnaE), encoding the
a
subunit of pol III, was required for lesion bypass in vivo.
This led to the incorrect model that the UmuC and
UmuD
0
proteins were accessory proteins, which enabled
pol III to bypass DNA lesions. Finally, UmuCD
0
-
dependent lesion bypass systems were reconstituted
Encyclopedia of Biological Chemistry, Volume 4. q 2004, Elsevier Inc. All Rights Reserved. 308
from purified components, and led to the finding that
UmuC has DNA polymerase activity, and together with
UmuD
0
it forms pol V. This discovery was made
simultaneously and independently in the laboratories
of Myron Goodman (University of Southern California,
Los Angeles) and Zvi Livneh (Weizmann Institute of
Science, Rehovot, Israel).
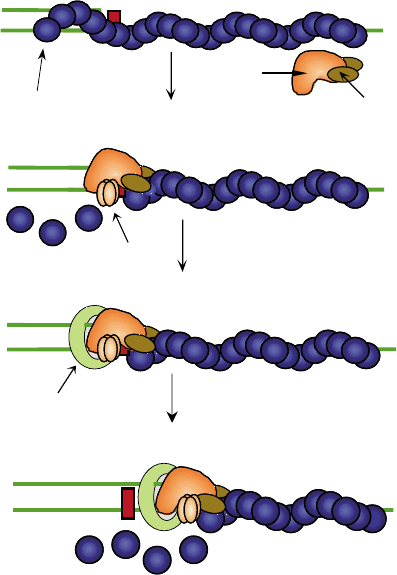
Model for the Mechanism of Lesion
Bypass by Pol V
Pol V is unable to bypass lesions on its own. It requires
two additional proteins, RecA and SSB, known to be
involved in other DNA transactions such as replication
and recombination. In addition, pol V is stimulated by
the processivity proteins, which are also required for
DNA replication. The RecA protein is required as a
nucleoprotein complex with ssDNA, in which multiple
RecA monomers form a protective helical sleeve around
the DNA (Figure 1, stage 1). The RecA filament prevents
nonproductive binding of pol V to ssDNA regions
remote to the primer terminus, and it targets pol V to
the primer-template region (Figure 1,stage2).
Once targeted to the primer-terminus site, the RecA
filament needs to locally dissociate, to allow productive
binding of pol V. This requires, in addition to pol V, also
the SSB protein. The pol V initiation complex is
stabilized by RecA–UmuD
0
interactions, UmuC– DNA
interactions, and perhaps interactions with SSB. Once
this pol V complex has formed an initiation complex,
DNA synthesis can occur. However, to get full pol V
holoenzyme activity, the processivity proteins are
needed, including the
b
-subunit DNA sliding clamp,
and the
g
-complex clamp loader (Figure 1, stages 3, 4).
These proteins are essential for highly processive DNA
replication by DNA polymerase III; however, they also
increase the processivity of DNA polymerases II, IV, and
V, although to a much lesser extent than pol III. DNA
synthesis by pol V holoenzyme causes dissociation of
RecA monomers, as the polymerase progresses. Pol V is
a major lesion-bypass machine, which can bypass a wide
variety of DNA lesions. This includes abasic sites, and
the two major lesions caused by UV light, a cyclobutyl
thymine– thymine dimer, and a thymine–thymine 6–4
adduct. Most remarkably, pol V is able to bypass a
stretch of 12 methylene residues inserted into the DNA.
This artificial lesion is an extreme form of DNA damage,
since it bears no resemblance to any of the regular DNA
constitutents. Despite this remarkable bypass ability, pol
V is poor in bypassing some DNA lesions, most notably
benzo[a]pyrene–guanine adducts. The ability of pol V
to bypass DNA lesions correlates with its low fidelity.
However, low fidelity by itself is not sufficient to ensure
effective lesion bypass, since there are enzymes with
fidelity similar to pol V, which nevertheless are not
lesion-bypass enzymes (e.g., mammalian DNA poly-
merase
b
). The crystal structure of pol V was not
determined yet. However, based on several crystal
structures of other homologous lesion-bypass DNA
polymerases, a spacious and flexible active site is likely
to be responsible, at least in part, for the ability to bypass
DNA lesions.
The Fidelity of Pol V
The fidelity of DNA synthesis by pol V undamaged DNA
templates is 10
23
–10
24
errors/nucleotide replicated.
This is 100–1000-fold lower than the fidelity of the
replicative pol III holoenzyme. DNA sequence analysis
of pol V errors showed that pol V produces all types of
errors, including frameshifts and base substitutions.
However, it has a propensity to produce transversion
mutations, namely, changing a pyrimidine into a purine,
or vice versa. Pol V forms transversions at a frequency
up to 300-fold higher than pol III holoenzyme (which
tends to form transition mutations; Table I). It should be
4. Lesion bypass
M
UmuC
UmuD'
1. Formation of a RecA-ssDNA nucleoprotein filament
Pol V
RecA
2. Targeting of pol V to the primer terminus
SSB
3. Loading of the -subunit DNA sliding clamp
-subunit
β
β
FIGURE 1 Outline of the mechanism of lesion bypass by pol V. The
red square represents a blocking lesion. The “M” represents an incorrect
insertion opposite the lesion. Reproduced from Livneh, Z. (2001). DNA
damage control by novel DNA polymerases: Translesion replication
and mutagenesis. J. Biol. Chem. 276, 25639–25642, with permission
of the American Society for Biochemistry and Molecular Biology.
UmuC, D LESION BYPASS DNA POLYMERASE V 309

noted that base –base mismatches produced by DNA
polymerases during synthesis on undamaged DNA are
potential substrates for the mismatch repair system. This
system primarily corrects mismatches that are precur-
sors for transition mutations, namely, the types of
mismatches that the replicative pol III holoenzyme
tends to make. This significantly reduces mutations
that can occur during DNA replication, and contributes
to the high replication fidelity. The repair by the
mismatch repair system of mismatches that are pre-
cursors of transversions, such as pol V tends to make, is
20-fold less effective than transition mismatches. This
implies that pol V generates mismatches that are largely
immune to mismatch repair and yield mutations. This
provides the mechanistic basis for the phenomenon of
untargeted SOS mutagenesis, also termed SOS mutator
activity. In essence, this phenomenon involves the
formation of mutations in undamaged regions of DNA
in cells in which the SOS response was induced. This
process is umuDC- and recA- dependent, and primarily
yields transversion mutations. Based on the fidelity of
pol V, untargeted mutagenesis can be explained by the
activity of pol V under SOS conditions in undamaged
regions of the E. coli chromosome. It should be noted
that there is a second branch of untargeted mutagenesis
that depends on pol IV rather than pol V.
Regulation of Pol V
The activity of pol V is tightly regulated at several levels,
to ensure that this mutator polymerase acts only under
situations, and at sites, when it is really needed. As
described above, the umuDC operon is regulated at the
transcriptional level by the LexA repressor and the RecA
activator like other SOS genes. Within the timed
expression of SOS genes, the umuDC operon is induced
late, nearly an hour after the onset of induction. When
SOS genes become repressed again, as the SOS response
is shut down, the umuDC operon is repressed early. This
late induction, early shutdown expression pattern
provides an extra layer of regulation on pol V. It limits
the activity of pol V to a stage after error-free repair
mechanisms had a chance to repair DNA, and even at
that late stage in SOS, the activity of pol V is limited in
time, as it is shut off early. Additional regulation is at the
posttranslational level. The UmuD
2
protein is cleaved to
yield a shorter protein termed UmuD
0
2
, which is the
active protein in pol V. The proteolytic processing occurs
via the self-cleavage of the UmuD
2
protein, promoted by
activated RecA, in a mechanism that is similar to the
cleavage of the LexA repressor. Finally, there is
regulation by turnover, as UmuD is degraded by the
Lon and ClpXP proteases. The steady-state level of
UmuC in noninduced cells is low and undetected by
currently available antibodies. Upon SOS induction,
UmuC accumulates to a level of , 200 molecules/cell.
UmuD is present in higher amounts, starting with 200
molecules in the noninduced cell, and going up to 2400
molecules/cell after induction. Finally, pol V is regulated
at the activity level, in its requirement for a RecA
nucleoprotein filament. Such filaments are formed in
DNA only when replication is arrested (e.g., at a lesion)
and a persistent single-stranded region is generated.
In vivo Function of Pol V
The major phenotype of E. coli cells lacking pol V is a
strong decrease in mutations caused by DNA-damaging
agents such as UV radiation or MMS. Survival is
decreased too, but to a lower degree compared to
defects in other DNA repair pathways, such as excision
repair. Based on these phenotypes, there are two views
on the in vivo role of pol V. One is that pol V repairs
replication gaps and this leads to increased survival.
According to this model, the mutations formed by pol V
are a by-product of the gap-filling activity, and can be
viewed as the price paid for this gap repair (repair at the
expense of mutations). The other view portrays the
generation of mutations as the main role of pol V.
According to this view, when a population of E. coli
cells is under hostile environmental conditions, e.g., lack
of nutrients, or presence of DNA-damaging agents such
as UV light, it increases its mutations rate via pol V, to
facilitate adaptation and increase fitness. Even one
successful mutation in a cell (e.g., conferring higher
resistance to sunlight) may have a great impact on the
TABLE I
Mutations Produced during In Vitro DNA Synthesis by Pol V and
Pol III Holoenzyme
Mutation
frequency 3 10
25
/gene
Mutation Mismatch
a
Pol III Pol V Pol V/pol III
Transition
T ! CT:G, 1.4 368.3 . 263
Transversion
A ! C A:G 2.8 161.1 58
A ! T A:A , 1.4 414.4 . 296
C ! A C:T , 1.4 92.1 . 66
T ! A T:T 2.8 230.2 82
T ! GT:C, 1.4 69.1 . 49
a
The mutations were formed in the cro repressor gene of phage
l
during gap-filling DNA synthesis.
Reproduced from Maor-Shoshani, A., Reuven, N. B., Tomer, G.,
and Liuneb, Z. (2000). Highly mutagenic replication by DNA
polymerase V (UmuC) provides a mechanistic basis for SOS untargeted
mutagenesis. Proc. Natl Acad. Sci. USA 97, 565–570, with permission
of the National Academy of Sciences, U.S.A.
310 UmuC, D LESION BYPASS DNA POLYMERASE V
entire population, since in time these cells, which have a
selective advantage, may proliferate to a state where
they take over the population. Consistent with such an
idea is the finding that when mutator strains are grown
for many generations along with nonmutator strains, the
mutator strain takes over. Similarly, umuDC-deficient
mutants suffer a severe reduction in fitness when
competing against cells with normal pol V. Since
mutations form randomly in relation to their functional
outcome, a mutator acting long enough will start
accumulating deleterious mutations, and will lose its
adaptive advantage. The fact that the activity of pol V is
transient (namely, as long as the SOS response is on)
helps to preserve successful mutations from being
neutralized by the formation of other mutations. It
was also suggested that pol V initially evolved as a
generic bypass DNA polymerse, which functioned to
increase survival by overcoming replication blocks, but
as more sophisticated DNA repair mechanisms evolved,
the importance of the bypass function decreased, and
what kept pol V during evolution was its mutator effect.
Of course, the various hypotheses are not mutually
exclusive, and the in vivo role of pol V may be both in
survival and mutagenesis.
Bacterial Homologues of Pol V
E. coli contains a homologue of pol V, termed pol IV,
which is the product of the SOS gene dinB. Like pol V,
pol IV is an SOS-inducible, low-fidelity, and low-
processivity DNA polymerase, which is able to bypass
some DNA lesions. As in pol V, the processivity and
lesion-bypass ability of pol IV are increased by the
b
-subunit clamp, and the
g
-subunit clamp loader.
However, unlike pol V, pol IV is a single subunit
enzyme, and its bypass activity requires neither RecA
nor SSB. On undamaged DNA, pol IV tends to form
frameshifts, primarily minus-one deletions. The in vivo
role of pol IV is not as clear as that of pol V. Pol IV is
required for untargeted mutagenesis of phage
l
. This
mutagenic pathway is observed in nonirradiated phage
l
when it infects a UV-irradiated E. coli host. In addition,
pol IV is required for the bypass of a benzo[a]pyrene–G
adducts. It is also required for stationary phase
mutations.
Homologues of pol V exist in many, but not all
bacteria. Pol V homologues were also found on native
bacterial conjugative plasmids, such as R46. These are
large plasmids, in the range of 100 kbp, with broad host-
range specificity, which often carry multiple antibiotics-
resistance markers. In the case of the R46 plasmid, the
UmuD, UmuD
0
, and UmuC homologues are MucA,
MucA
0
and MucB, respectively. MucA
0
and MucB were
shown to form a lesion-bypass DNA polymerase, pol RI,
with properties similar to those of pol V. Pol RI,
like pol V, required both RecA and SSB for bypassing
an abasic site. MucA and MucB are present on plasmid
pKM101, a natural deletion derivative of plasmid R46.
Plasmid pKM101 was introduced into the Salmonella
strains that are used in the Ames test for mutagens, in
order to improve its mutagenic sensitivity.
The emergence of pathogenic bacteria that are
resistant to multiple types of antibiotics is associated in
part of the cases with the presence of native conjugative
plasmids. The fact that native plasmids, which have a
limited genome size, and rely heavily on host factors,
carry mutator lesion-bypass DNA polymerase genes
indicates that the latter have a special role in the life
cycle of such plasmid. One possibility is that the mutator
activity of the polymerases is required when the
plasmids enter a new foreign bacterial host, and need
rapid adaptation to the new intracellular environment.
Eukaryotic Homologues of Pol V
Homologues of pol V were found to be conserved in
evolution, and comprise the Y family of DNA poly-
merases. The yeast S. cerevisiae contains two homol-
ogues of UmuC, pol
h
(product of the RAD30 gene), and
REV1, a G-template specific DNA polymerase. Human
cells contain no less than four homologues of pol V: pol
h
, pol
i
, pol
k
, and REV1. The DNA polymerases of
the Y family were discovered in 1999 by several
investigators, simultaneously and independently.
The S. cerevisiae pol
h
was reported first, discovered
by Satya Prakash and Louis Prakash (University of Texas
Medical Branch, Galveston). Both the S. cerevisiae and
the human pol
h
have the remarkable property that they
replicate across an unmodified DNA TT sequence, with
similar efficiency and specificity to that of a TT
cyclobutyl dimer. In the absence of pol
h
in the cell,
another polymerase takes over, but performs the
reaction with a lower fidelity, leading to increased UV
mutagenesis. This is the situation in the human
hereditary disease Xeroderma Pigmentosum Variant
(XP-V). Unlike other forms of XP, which are defective
in components of the error-free nucleotide excision
repair, XP-V patients are deficient in DNA pol
h
(the
XP-V gene product). XP-V patients lacking this enzyme
show extreme sensitivity to sunlight and a high
susceptibility to skin cancer. This led to the unexpected
conclusion that lesion bypass may be functionally
nonmutagenic, and that lesion-bypass DNA poly-
merases may function to protect mammals from at
least certain types of cancer.
S. cervisiae has an additional lesion-bypass polymer-
ase, pol
z
, encoded by the REV3 and REV7 genes. It is
homologous to the replicative pol
d
, and not to the
Y-family DNA polymerases. Historically, pol
z
was the
first lesion-bypass DNA polymerase, discovered in 1996
UmuC, D LESION BYPASS DNA POLYMERASE V 311
by David Hinkle and Christopher Lawrence (Rochester
University), followed soon thereafter by the discovery, in
the same year, and by the same investigators, that REV1
has nucleotidyl transferase activity. Human cells also
contain additional polymerases, which can bypass
lesions in vitro. This includes pol
m
and pol
l
,and
homologues of the yeast REV3 and REV7 genes,
encoding a putative pol
z
. Overall this suggests that
lesion bypass has an important role in the response to
DNA damage in mammalian cells.
SEE ALSO THE FOLLOWING ARTICLES
DNA Base Excision Repair † DNA Damage:
Alkylation † DNA Polymerase I, Bacterial † DNA
Polymerase II, Bacterial † DNA Polymerase III,
Bacterial † Translesion DNA Polymerases, Eukaryotic
GLOSSARY
base substitution mutation A mutation in which a particular DNA
base is changed to another base (e.g., A ! G).
benzo[a]pyrene–guanine adduct A DNA adduct formed between
benzo[a]pyrene, a major tobacco smoke carcinogen, and a guanine
base. Benzo[a]pyrene requires metabolic activation before it can
react with DNA.
excision repair The major mechanism of DNA repair. It involves
excision of the damaged site from DNA, followed by gapfilling
DNA synthesis, based on the complementary intact strand and
ligation.
frameshift mutation A mutation that changes the reading frame of a
gene. Frameshifts involve either deletion or insertion of one or two
nucleotides. Larger deletions or insertions may also cause frame-
shift mutations, when the number of deleted or added nucleotides is
not a multiple integer of three.
operon A segment of the genome with two or more genes which are
cotranscribed yielding a multicstronic mRNA.
processivity of DNA polymerase The number of nucleotides poly-
merized by a DNA polymerase per single binding event to the
primer-template. Processivity of DNA polymerases varies from 1
(distributive DNA polymerase) up to 10 000 (highly processive
DNA polymerase). Usually replicative polymerases have very
high processivity, whereas DNA repair polymerases have lower
processivity.
regulon A set of genes, residing at different locations in the
chromosome, that are regulated by a common regulatory pathway.
The genes regulated by the SOS response in E. coli comprise a
regulon.
transition mutation A mutation in which a pyrimidine is changed to
another pyrimidine, or a purine is changed to another purine.
Changes of A ! GorC! T are examples of transition mutations.
transversion mutation A mutation in which a purine changes to a
pyrimidine, or a pyrimidine changes into a purine. Changes of
A ! CorT! A are examples of transversion mutations.
FURTHER READING
Baynton, K., and Fuchs, R. P. (2000). Lesions in DNA: Hurdles for
polymerases. Trends Biochem. Sci. 25, 74–79.
Goodman, M. F. (2000). Coping with replication ‘train wrecks’ in
Escherichia coli using Pol V, Pol II, and RecA proteins. Trends
Biochem. Sci. 25, 189– 195.
Livneh, Z. (2001). DNA damage control by novel DNA polymerases:
Translesion replication and mutagenesis. J. Biol. Chem. 276,
25639–25642.
Radman, M. (1999). Enzymes of evolutionary change. Nature 401,
866–869.
Sutton, M. D., Smith, B. T., Godoy, V. G., and Walker, G. C. (2000).
The SOS response: Recent insights into umuDC-dependent
mutagenesis and DNA damage tolerance. Annu. Rev. Genet. 34,
479–497.
BIOGRAPHY
Zvi Livneh received his Ph.D. from the Weizmann Institute of Science
in Israel, and was a postdoctoral fellow at Stanford University School
of Medicine in California. He is a Professor of biochemistry and
molecular medicine, and Head of the Department of Biological
Chemistry at the Weizmann Institute of Science in Israel. His main
research interests are molecular mechanisms of DNA repair and
mutagenesis in bacteria and in mammals, and the role of DNA repair in
cancer risk.
312 UmuC, D LESION BYPASS DNA POLYMERASE V