Lennarz W.J., Lane M.D. (eds.) Encyclopedia of Biological Chemistry. Four-Volume Set . V. 4
Подождите немного. Документ загружается.


Uncoupling Proteins
Daniel Ricquier and Fre
´
de
´
ric Bouillaud
Centre National de la Recherche Scientifique, Paris, France
Uncoupling proteins (UCPs) are mitochondrial transporters
present in the inner membrane of mitochondria. They belong
to the family of anion mitochondrial carriers including adenine
nucleotide transporters, phosphate carrier and other transpor-
ters. The term uncoupling protein was originally given to
UCP1 which is uniquely present in mitochondria of brown
adipocytes, the thermogenic cells devoted to maintenance of
body temperature in mammals. In these cells, UCP1 acts as a
proton carrier creating a shunt between complexes of
respiratory chain and the ATP-synthase. Purine nucleotide
inhibit UCP1 whereas fatty acids activate it. Activation of
UCP1 stimulate respiration and the uncoupling process results
in a futile cycle and dissipation of oxidation energy into heat.
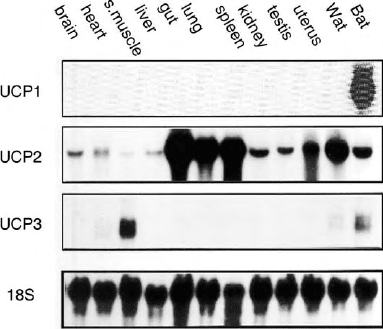
UCP2 is ubiquitous and highly expressed in lymphoid system
and macrophages. UCP3 is mainly expressed in skeletal
muscles. In comparison to the established uncoupling and
thermogenic activities of UCP1, UCP2, and UCP3 rather
appear to be involved in the limitation of free radicals levels in
cells than in physiological uncoupling and thermogenesis. The
mechanism of the protonophoric activity of the UCPs is still
controversial since it has been proposed that the UCPs
transport fatty acid anions and catalyse proton transport by
fatty acid cycling. Quinones and superoxide ions may also
activate the UCPs.
The Mitochondria and the
Coupling of Respiration
to ADP Phosphorylation
Cellular respiration, the reactions of the citric acid cycle,
fatty acid oxidation, and several steps of urea synthesis
and gluconeogenesis take place in specialized cellular
organites, the mitochondria.
MITOCHONDRIA
In addition to oxidative phosphorylation and metabolic
pathways, mitochondria are involved in thermogenesis,
radical production, calcium homeostasis, apoptosis, and
protein synthesis. Mitochondria contain two compart-
ments bounded by inner and outer membranes. The
outer membrane is permeable to many small metabolites
whereas the permeability of the inner membrane is
controlled in order to maintain the high electrochemical
gradient created by the mitochondrial respiratory chain
which is necessary for energy conservation and ATP
synthesis in mitochondria. The inner membrane trans-
ports anion substrates such as ADP, ATP, phosphate,
oxoglutarate, citrate, glutamate, and malate.
COUPLING OF RESPIRATION
TO
ATP SYNTHESIS
It has long been known that respiration and mitochon-
drial ATP synthesis are coupled. The observation that
respiration rate increased when mitochondria syn-
thesized more ATP led to the concept of respiratory
control by ADP phosphorylation. In fact, there is a link
between mitochondrial ATP synthesis and cellular ATP
demand by a feed-back mechanism. In agreement with
Mitchell’s theory, it was demonstrated that the mito-
chondrial electrochemical proton gradient, generated as
electrons are passed down the respiratory chain, is the
primary source for cellular ATP synthesis. In this way,
several complexes of the respiratory chain pump protons
outside of the inner membrane during reoxidation of
coenzymes and generate a proton gradient which is
consumed by the reactions of ATP synthesis. Proton leak
represents another mechanism consuming the mito-
chondrial proton gradient. Mitchell’s theory predicted
that any proton leak non coupled to ATP synthesis
would represent an uncoupling of respiration and result
in thermogenesis. An excellent example of such an
uncoupling of respiration to ADP phosphorylation is
represented by the mitochondrial UCP of brown
adipocytes (UCP1) which dissipates energy of substrate
oxidation as heat.
Brown Adipose Tissue and UCP1:
History of a True
Respiration Uncoupling
Maintenance of body temperature in a cold environment
or at birth requires thermogenesis. Another situation
Encyclopedia of Biological Chemistry, Volume 4. q 2004, Elsevier Inc. All Rights Reserved. 313
requiring thermogenesis is arousal from hibernation.
The two regulatory thermogenic processes are shivering
and metabolic thermogenesis also referred to as non-
shivering thermogenesis. The largest part of nonshiver-
ing thermogenesis in small mammals is achieved in the
brown adipose tissue (BAT).
BROWN ADIPOSE TISSUE (BAT)
BAT is found in all small mammals and in the newborn
of larger mammals, such as humans. BAT is located in
specific body areas near large blood vessels and consists
of brown adipocytes which are distinct from white
adipocytes. The brown adipocytes are characterized by
numerous mitochondria containing a highly developed
inner membrane (Figure 1). Activation of thermogenesis
in BAT occurs in newborns, rodents exposed to cold, and
in animals emerging from hibernation. It is commanded
by the central nervous system and the orthosympathetic
fibers innervating each brown adipocyte. The norepi-
nephrine released by these fibers binds to adrenergic
receptors on the surface of the brown adipocytes. The
later steps of the activation of thermogenesis in brown
adipocytes are production of cyclic AMP, activation of
lipolysis, and oxidation of fatty acids by the numerous
mitochondria. Released fatty acids stimulate brown
adipocyte respiration and heat production.
RESPIRATION UNCOUPLING IS THE
THERMOGENIC MECHANISM
IN
BROWN ADIPOCYTES
In most cell types such as muscular fibers, ATP
utilization stimulates ADP phosphorylation and respir-
ation (Figure 2). Original observations made in 1967
revealed that the high respiratory rate in brown
adipocyte mitochondria was not controlled by mito-
chondrial ADP phosphorylation suggesting that energy
from substrate oxidation was dissipated into heat
instead of being converted in ATP (Figure 2). In line
with Mitchell’s chemiosmotic theory, if proton leakage
is activated by a signal such as free fatty acids in the
case of the mitochondria of brown adipocytes, the
mitochondrial membrane potential is decreased and
concomitantly, respiration is activated. Since, in
addition, the activated proton leakage is not linked
to ADP phosphorylation, respiration is uncoupled
from ATP synthesis and oxidation energy is dissipated
in the form of heat.
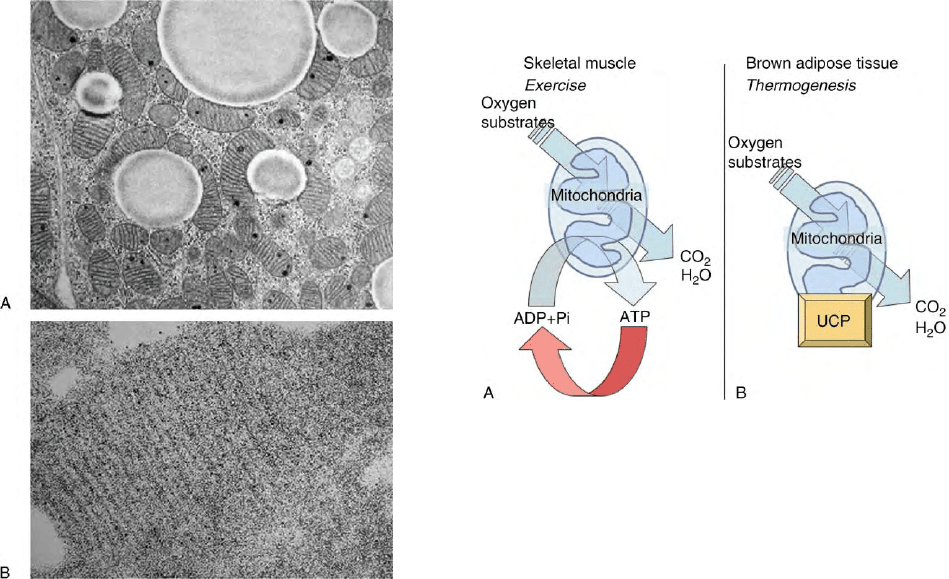
FIGURE 1 Histology of brown adipocyte. (A) The cytosol of brown
adipocytes is characterized by numerous mitochondria and lipid
droplets. (B) Magnification of a brown adipocyte mitochondrion
showing parallel cristae and UCP1 detected using antibodies (black
dots). (Figure kindly provided by Dr Saverio Cinti, University of
Ancona.)
FIGURE 2 Comparison of skeletal muscle and brown adipose tissue
mitochondria. (A) Mitochondria are cellular organelles converting the
redox energy liberated during oxidation of organic substrates into ATP,
a molecule containing energy under a form readily usable by most
enzymes working to maintain cell structure and integrity or to perform
work such as mechanical work in muscle. The cellular need for ATP
controls oxidation rate by mitochondria. (B) In brown adipocyte, a
specific uncoupling protein referred to as UCP (precisely UCP1) is
present in a large amount in the inner membrane. UCP1 is able to fool
mitochondria which accelerates their oxidation rate whereas no ATP is
produced. UCP1 removes the control of energy expenditure by ATP
demand and therefore oxidation rate and energy expenditure increase
under the form of heat. This process is used in newborns, arousal from
hibernation, and small mammals exposed to cold when muscle
thermogenesis would not be sufficient.
314 UNCOUPLING PROTEINS
THE UCP OF BROWN
ADIPOCYTE MITOCHONDRIA
The protein responsible for proton leakage within the
inner mitochondrial membrane and for respiratory
uncoupling was identified in 1976–77, was purified,
and its cDNA was cloned. It has an apparent molecular
weight of 33 000 and was originally called UCP. This
protein is known as UCP1 since the discovery of UCP2 in
1997. UCP1 is abundant in the inner membrane of the
mitochondria of brown adipocytes and is specific to
these cells. It short-circuits the proton circuit between
complexes of the respiratory chain and the mitochon-
drial ATP-synthase which is the main consumer of the
proton gradient (Figure 3). When there is no need for
thermogenesis, purine nucleotides bound to UCP1
inhibit its activity. Upon activation of thermogenesis
by the sympathetic nervous system, fatty acids overcome
the inhibitory effect of nucleotides and activate UCP1.
Heterologous expression of UCP1 in yeasts or mamma-
lian cell lines induces respiration uncoupling. The
protonophoric activity of UCP1 has been reconstituted
in liposomes where it can be inhibited by nucleotides and
activated by free fatty acids. The mechanism of action of
UCP1 is still controversial: some scientists believe that it
is a true proton transporter, whilst others assert that it
returns anionic fatty acids to the intermembrane space,
after they have crossed the membrane in protonated form.
The observation that Ucp12 /2 mice were unable
to maintain body temperature in the cold proved that
the UCP1-induced uncoupling of respiration was
responsible for cold-activated nonshivering thermogen-
esis of rodents. In agreement with this observation,
ectopic expression of UCP1 in skeletal muscles of
transgenic mice promotes substrate oxidation in muscles
and resistance to obesity and type 2 diabetes. UCP1
functions as a dimer and each monomer is made of six
transmembrane fragments (Figure 4). Moreover, UCP1,
the ADP/ATP translocator and other mitochondrial
anion carriers derive from the same ancestral gene and
probably share a similar structure. They all have a
triplicated structure (Figure 4).
The Novel UCPs
Given their specific role in thermogenesis, it has always
seemed logical that brown adipocytes be equipped with
an original mechanism, partial uncoupling of respir-
ation, brought into play by a specific protein, UCP1,
which induces proton leakage. In fact, it is known that
mitochondrial respiration is always accompanied by
heat production as it is imperfectly coupled to ADP
phosphorylation in all types of cells. To explain this
incomplete coupling of respiration and the energy loss
mechanism, some authors have invoked slippage of the
respiratory chains, while others referred to proton leaks.
It has been estimated that proton leaks from the
inner membrane of mitochondria of hepatocytes
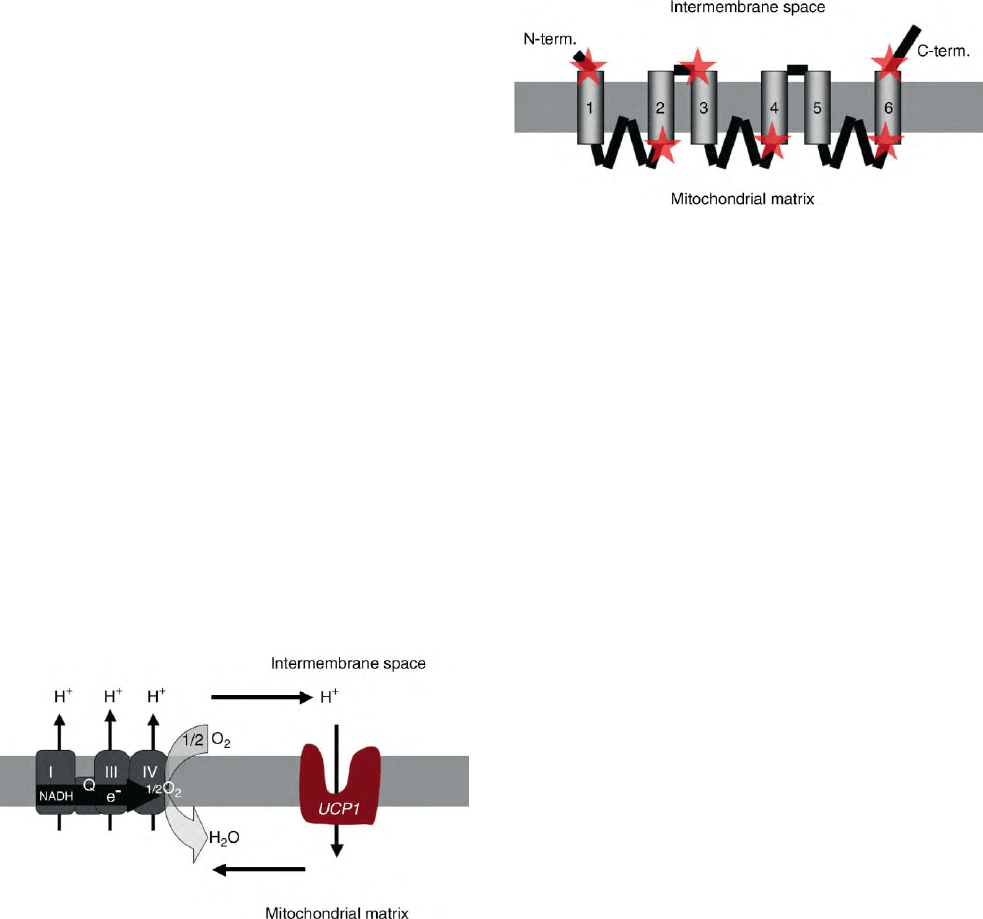
FIGURE 4 Model of insertion of UCP in the membrane. The
determination of structure of the UCPs remains difficult because
crystallization of such membrane proteins has not been successful yet.
To modelize the organization of such proteins, indirect methods such
as prediction of secondary structure by computers can be used.
Experimentally, restricted domains of the protein in the membrane
(indicated by stars) were explored using specific antibodies. The UCPs
are made of six transmembraneous segments (probably alpha helices),
and three hydrophilic loops stemming from the matricial side of the
membrane. An internal repetition in the structural arrangement of the
protein suggests that the protein (made of 300 amino acid residues)
derived from the ancient triplication and divergence of a 100 amino
acid domain (made of 2 helices and 1 loop).
FIGURE 3 UCP1-induced proton cycling. Q (coenzyme Q) refers to
complex II which is the succinate dehydrogenase. This enzyme directly
reduces coenzyme Q but has no proton pumping activity contrary to
complexes I, III and IV. UCP is inserted in the mitochondrial inner
membrane where, also present, is a multienzymatic complex called the
respiratory chain made of complexes I to IV. The respiratory chain
reoxidize reduced coenzymes and electrons are driven to oxygen. This
oxido-reduction step liberates energy which is used to generate an
electrochemical gradient of protons across the inner membrane. This
gradient is normally consumed by the ATP-synthase which phosphor-
ylates ADP. UCP1 transports protons passively and makes possible a
futile cycle of protons across the inner membrane leading to increased
energy expenditure. This schema illustrates the situation encountered
in brown adipocytes of mammals where a large amount of respiratory
chains as well as a large amount of UCP1 are present. Activation of the
futile cycling increases considerably energy expenditure and thus heat
production in these thermogenic cells. In other cells where homologues
of the UCP1 are expressed at much lower level, this pathway would
represent a minor contributor to energy expenditure, but might be of
importance to avoid oxidative damage.
UNCOUPLING PROTEINS 315
and myocytes could explain about 20% of the body’s
basal metabolism.
UCP2: A HOMOLOGUE OF THE BROWN
FAT UCP PRESENT IN VARIOUS TISSUES
A clone corresponding to a protein with 59% sequence
identity with the UCP of brown fat was isolated. This
new UCP, called UCP2, was also identified in humans. In
contrast with UCP1, the mRNA of UCP2 is present in
almost all tissues and many cell types, such as spleen,
thymus, digestive tract, lung, brain, adipose tissues,
skeletal muscle, heart, adipocytes, myocytes, and
macrophages (Figure 5). Expressed in yeasts, murine
UCP2 decreased the potential of the mitochondrial
membrane, raised the respiration rate and reduced
sensitivity to uncouplers: UCP2 therefore was able to
uncouple respiration and appeared to be a second
mitochondrial UCP. The UCP2 gene is located on
chromosome 7 of the mouse and chromosome 11 of
humans, near a region linked to hyperinsulism and
obesity. The expression of UCP2 mRNA was measured
in obesity-prone and obesity-resistant mice given a lipid-
rich diet: the obesity-resistant mice overexpressed UCP2
mRNA in their adipose tissue. These findings, together
with the function of the protein and the chromosomal
location of its gene, led to propose a role for UCP2 in
diet-induced thermogenesis. However, this hypothesis
was not validated in Ucp22 /2 mice.
UCP3: ANOTHER UCP HOMOLOGUE
PREDOMINANTLY PRESENT
IN
SKELETAL MUSCLE
cDNAs corresponding to UCP3, a protein homologous
to UCP1 and UCP2, were cloned soon after the
discovery of UCP2. The amino acid sequence of UCP3
is 72% identical to that of UCP2 and 57% identical to
that of UCP1. UCP3 mRNA is predominantly expressed
in the skeletal muscles of humans, mice, and rats
(Figure 5). Human and murine UCP2 and UCP3 genes
are juxtaposed on the same chromosome.
PLANT AND BIRD UCPS
Functional studies of isolated plant mitochondria
suggested that plants contain mitochondrial UCPs.
Following this observation, a cDNA from a new UCP
was isolated from a potato cDNA library. The sequence
of the corresponding protein, called stUCP, is 44%
identical to that of UCP1 and 47% identical to that of
UCP2. The mRNA of stUCP is present in most plant
organs. The most surprising result was that stUCP
mRNA was markedly induced in the leaves of plants
exposed to a temperature of 48C. Cold, therefore,
induced stUCP as it induces UCP1 in the brown adipose
tissue of animals. However, plant and animal UCPs may
have different functions. More recently, a chicken UCP
termed avUCP was characterized. This gene is only
expressed in skeletal muscles of birds and ducks and is
induced upon exposure to the cold. Therefore avUCP
seems to be involved in regulatory thermogenesis.
Other proteins referred to as UCP4 and BMCP1/UCP5
were characterized. However their exact relationship
to UCP1, UCP2, and UCP3 will require further work.
ROLE AND FUNCTION OF UCPSOTHER
THAN
UCP1: A ROLE IN CONTROLLING
THE
LEVEL OF REACTIVE
OXYGEN SPECIES
Numerous questions remain unanswered, notably con-
cerning the exact catalytic activity of each UCP, and the
nature of the endogenous ligands of UCP2 and UCP3.
These proteins may simply translocate protons. As said
above for UCP1, it has been proposed that all the UCPs
are active as fatty acid cycler through the membrane.
According to this later hypothesis, protonated fatty
acids cross the membrane and release a proton on the
matricial side; then, the UCP facilitates the translocation
of the anionic form of fatty acids. High loads of UCP2 or
UCP3 can uncouple respiration in yeast or mammalian
cell, but it is not certain whether the low physiological
levels of these proteins may be sufficient to induce a net
uncoupling of respiration in vivo.Thedivergent
observations of the regulation of the activities of UCP2
or UCP3 by fatty acids or nucleotides will require further
studies. Interestingly, it has been proposed that UCP2
and UCP3 could export fatty acid anion under con-
ditions of elevated fatty acid oxidation.
FIGURE 5 Tissular distribution of UCP1, UCP2, and UCP3 RNAs.
The data correspond to mouse tissues. A very similar picture was
obtained when analyzing human tissues. 18S refers to ribosomal RNA.
316 UNCOUPLING PROTEINS
Metabolic Activity of UCP2 and UCP3
Conflicting data regarding the association of UCP2 or
UCP3 genetic polymorphisms to body mass index,
susceptibility to obesity, resting metabolic rate, meta-
bolic efficiency, fat oxidation, insulin resistance, suscep-
tibility to gain fat with age were obtained. However,
UCP2 appears to act as a negative regulator of insulin
secretion. Moreover, mice overexpressing a large
amount of human UCP3 in skeletal muscle weigh less,
have a decreased amount of adipose tissue, and an
increased resting oxygen consumption.
Reactive Oxygen Species
Guided by the fact that UCP2 is expressed at high level
in the immune system, a role for UCP2 in immunity was
searched for. In fact, Ucp2 2 /2 mice are more resistant
to infection by a parasite. This phenotype was explained
by the fact that disruption of the Ucp2 gene provoked an
elevation of ROS level that facilitated the killing of the
pathogens. Mice lacking the Ucp3 gene also produced
more oxygen radicals in myocytes. Therefore, it appears
that UCP2 and UCP3 contribute to limit the level of
reactive oxygen species in cells. Such an activity may be
explained by a mild uncoupling activity of the proteins
since a small decrease of the mitochondrial membrane
potential opposes production of superoxide ion in
mitochondria (Figure 6). The observation that UCP2
protects against atherosclerosis highlights its ability to
counteract radical synthesis.
UCP1, UCP2, and UCP3,
Conclusions and Perspectives
UCP1 has a well-demonstrated uncoupling activity and
an essential role in maintenance of body temperature in
small rodents exposed to the cold, but the exact
biochemical and physiological roles of UCP2 and
UCP3 remain to be further identified. Certain data
support a true activity of these UCPs in respiration
uncoupling and substrate oxidation, other data do not
agree with a role for these UCPs in controlling adiposity.
Several reports support a role for UCP2 and UCP3 in
decreasing the level of reactive oxygen species in cells.
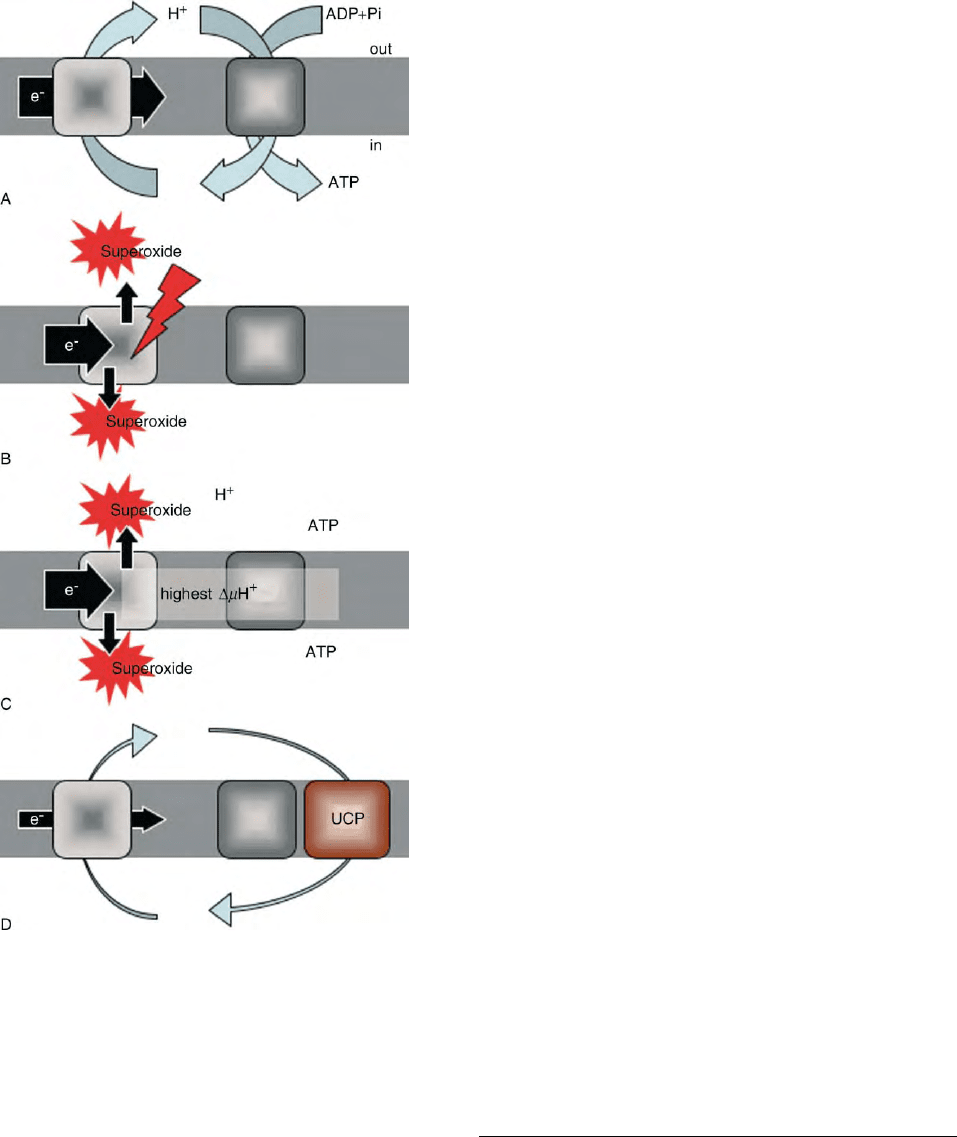
FIGURE 6 Role of UCP and superoxide generation by mitochondria.
(A) mitochondria phosphorylating ATP: a proton gradient generator
(the respiratory chain, left square) uses the energy liberated by the
movement of electrons to pump protons through the membrane (grey
zone) towards the intermembrane space. A proton gradient consumer
(the ATP-synthase, right square) phosphorylates ADP coming from
outside into ATP, that is exported afterwards. Therefore, a proton cycle
across the inner membrane couples oxidation to ATP synthesis (and
vice versa). (B) and (C) when oxidation is impaired (B) or slows down
(C), an excess of electrons aiming to travel across the respiratory
complexes cannot reach oxygen (to form water). Single electrons react
with molecular oxygen and form superoxide anion (O2
z2
). It occurs
either when the respiratory chain is poisoned (red arrow in panel B) or
under physiological circumstances when the use of ATP by the cell is
weak, ATP level rises, and ADP level decreases (panel C). In that case,
the membrane potential rises to its highest value which impedes proton
pumping, slows down reoxidation, and therefore increases superoxide
production. (D) in tissues other than the thermogenic brown adipose
tissue, the UCPs may induce a controlled leak of protons across the
inner membrane that allows reoxidation to occur and maintain the
membrane potential below the threshold inducing superoxide gener-
ation. The mechanism of proton transport (proton channeling or fatty
acid cycling), as well as the identity of regulators of UCPs inside the
cells are still controversial. Fatty acids would be cofactors of the proton
transport whereas nucleotides are inhibitors. The membrane potential
itself is a variable influencing heavily UCP1 activity. It has been
proposed that superoxide anion and quinones directly activate proton
transport by UCPs.
UNCOUPLING PROTEINS 317
Whilst UCP1 is present at a high level, an open question
is that of the exact amount of UCP2 and UCP3 in cells
since whatever is the exact intrinsic ability of these
proteins to uncouple respiration from ATP synthesis, a
certain amount of the proteins will be required to
achieve a physiological uncoupling. Finally, the possible
contribution of a UCP to the hypermetabolic syndrome
(known as Luft’s disease) previously described in
patients remains to be investigated.
SEE ALSO THE FOLLOWING ARTICLES
ATP Synthesis in Plant Mitochondria: Substrates,
Inhibitors, Uncouplers † Luft’s Disease † Mitochondrial
Membranes, Structural Organization † Quinones †
Respiratory Chain and ATP Synthase
GLOSSARY
mitochondria These organelles are the sites of respiration and
oxidative phosphorylation in all animal and higher plant tissues
as well as in protozoa, fungi, and aerobically grown yeasts. They
are , 2–30 mm long and 0.5–10 mm wide.
respiration coupling/uncoupling In coupled mitochondria, respira-
tion rate determines ADP phosphorylation rate and ATP synthesis
determines respiration rate. When uncoupling occurs (due to
chemical uncouplers or UCP), the respiration rate increases sharply
since the control by ADP phosphorylation does not limit respiration.
In such a situation, oxidation energy is dissipated as heat.
FURTHER READING
Boss, O., Hagen, T., and Lowell, B. B. (2000). Uncoupling proteins 2
and 3. Potential regulators of mitochondrial energy metabolism.
Diabetes 49, 143–156.
Cannon, B., and Nedergaard, J. (1985). The biochemistry of an ineffi-
cient tissue: Brown adipose tissue. Essays Biochem. 20, 110– 164.
Echtay, K. S., Winkler, E., and Klingenberg, M. (2000). Coenzyme Q is
an obligatory cofactor for uncoupling protein function. Nature
408, 609– 613.
Enerba
¨
ck, S., Jacobsson, A., Simpson, E. M., Guerra, C., Yamashita,
E. H., Harper, M. E., and Kozak, L. P. (1997). Mice lacking
mitochondrial uncoupling protein are cold-sensitive but not obese.
Nature 387, 90 –94.
Garlid, K. D., and Jaburek, M. (1998). The mechanism of proton
transport mediated by mitochondrial uncoupling proteins. FEBS
Lett. 438, 10 –14.
Himms-Hagen, J., and Ricquier, D. (1998). Brown adipose tissue. In
Handbook of Obesity (G. Bray, C. Bouchard and W. P. T. James,
eds.) pp. 415 –441. Marcel Dekker, New York.
Ledesma, M., Garcia
`
de Lacoba, M., and Rial, E. (2002). The
mitochondrial uncoupling proteins. Genome Biol. 3 (on line
November 29).
Nicholls, D. G., and Locke, R. M. (1984). Thermogenic mechanisms in
brown fat. Physiol. Rev. 64, 1–64.
Ricquier, D., and Bouillaud, F. (2000). The uncoupling protein
homologues: UCP1, UCP2, UCP3, StUCP & AtUCP. Biochem. J.
345(Pt 2), 161–719.
Skulachev, V. P. (1988). Uncoupling: New approaches to an old
problem of bioenergetics. Biochim. Biophys. Acta 1363, 100–124.
Stuart, J. A., Cadenas, S., Jekabsons, M. B., Roussel, D., and Brand,
M. D. (2001). Mitochondrial proton leak and the uncoupling
protein 1 homologues. Biochim. Biophys. Acta 1504, 144– 158.
BIOGRAPHY
Daniel Ricquier is Professor at Neckes-Enfants Malades Faculty of
Medicine and Fre
´
de
´
ric Bouillaud is Main Investigator at the Institut
National de la Sante
´
et de la Recherche Me
´
dicale. They work at CNRS
Unit 9078 at Faculte
´
de Me
´
decine and Institut de Recherches Necker-
Enfants Malades in Paris. They hold Ph.Ds from Pierre and Marie
Curie University in Paris. Their laboratory first cloned UCP1, UCP2,
BMCP1/UCP5, and avUCP. They also collaborated to the character-
ization of the first plant UCP. With their collaborators, they are authors
of original reports dealing with biochemistry, physiology, and genetics
of the UCP family.
318 UNCOUPLING PROTEINS

Unfolded Protein Responses
David Ron
New York University School of Medicine, New York, USA
The unfolded protein response (UPR) is a transcriptional and
translational response to the accumulation of unfolded proteins
in the endoplasmic reticulum (ER). The UPR is mediated by
highly conserved signaling pathways that are activated by
imbalance between the load of unfolded (or malfolded) ER
client proteins and the capacity of the organelle to process this
load. Collectively these pathways restore equilibrium to the
protein-folding environment in the organelle by increasing the
expression of genes that enhance nearly all aspects of ER
function and by transiently repressing the biosynthesis of new
client proteins. Interest in the UPR has been stimulated by the
realization that postsynthetic protein processing constitutes an
important step in gene expression and that protein malfolding
plays an important role in human disease.
ER Function and ER Stress
THE ER, A PROTEIN-PROCESSING
MACHINE
Proteins destined for secretion and membrane insertion
are translocated across the ER membrane in an unfolded
state. In the ER lumen they undergo chaperone-assisted
folding, a variety of organelle-specific post-translational
covalent modifications and often chaperone-assisted
assembly into oligomeric structures. Once properly
folded and assembled, most ER client proteins are
packaged into vesicles that are transported to more
distal sites in the secretory pathway. Proteins that fail to
attain their proper folded and oligomeric conformation
are retained in the ER by continued binding to
chaperones and are ultimately translocated from the
organelle to the cytoplasm for proteasomal degradation,
in a process known as ER-associated protein degra-
dation (ERAD).
To fulfill these various functions the ER is endowed
with a unique complement of proteins. These include
enzymes for post-translational covalent modifications
(e.g., N-linked glycosylation, disulfide bond formation),
chaperones that assist in folding and assembly steps,
translocation channels and transporters involved in
transmembrane traffic, and various components
involved in the postassembly steps of client protein
egress from the organelle. The build-up of these
components defines the capacity of the organelle to
handle its client proteins. In the late 1980s Sambrook,
Gething, and their colleagues discovered that manipula-
tions that interfere with the function of various aspects
of the ER client protein-handling machinery, and
thereby perturb protein folding in the ER, selectively
up-regulate the expression of genes that encode com-
ponents of that machinery. The extent of this transcrip-
tional response was not fully appreciated at the time,
however its selectivity was noted, hence its name: the ER
unfolded protein response (or UPR).
It was imagined that the cell possessed means to
monitor the load of client proteins presented to its ER
and responded to an increase in such load by up-
regulating the capacity of its ER to process unfolded
client proteins. An additional clue to the workings of this
response was provided by the seminal observation that
forced overexpression of an ER chaperone, BiP (also
known as GRP78) markedly suppressed the activity of
the UPR. Thus, at least one target gene of the UPR (BiP)
is able to exert negative feedback on the entire response.
BiP overexpression does not restore function to a
challenged ER; this requires the coordinate expression
of numerous UPR target genes. However, BiP, which is a
member of the highly conserved HSP70/DnaJ family of
chaperones, promiscuously recognizes hydrophobic
stretches of amino acids. These are normally incorpor-
ated into the cores of properly folded polypeptides and
assembled oligomeric complexes, but remain exposed
on the surface of unfolded, malfolded or unassembled
proteins. The ability of BiP to suppress the UPR
suggested that it might be doing so by masking a stress
signal generated by many different unfolded and
malfolded proteins. This phenomenon was assigned
the heuristic term ER stress and its level reflects the
balance between client protein load and the capacity of
the organelle to process that load.
PHYSIOLOGICAL AND
PATHOLOGICAL ER STRESS
To the extent that cell types vary in the load of
secreted proteins that they are called upon to produce,
Encyclopedia of Biological Chemistry, Volume 4. q 2004, Elsevier Inc. All Rights Reserved. 319
they are subject to widely different levels of physio-
logical ER stress. This explains enhanced activity of
the UPR in various professional secretory cells, such as
pancreatic cells of vertebrates or intestinal cells of the
nematode, Caenorhabditis elegans.ERstressalso
occurs when a mutation in an abundantly expressed
ER client protein renders that protein especially
difficult to fold. An example is provided by various
degenerative diseases affecting myelinated neurons in
which mutations in a component of the myelin sheath
(an ER client protein) cause the protein to malfold and
induce high levels of ER stress which, over time,
destroys the myelin-producing cell. It is important to
emphasize that most mutations that impede folding of
ER client proteins do not cause measurable ER stress.
However, because they diminish expression of the
properly folded protein, such mutations may deprive
the organism of the latter’s beneficial actions. This
genetic mechanism underlies such serious human
diseases as cystic fibrosis, familial hypercholesterol-
emia, and hemophilia. Nonetheless, the level of
expression of the mutant protein is not enough to
globally challenge ER function in the cell that produces
it and the phenotypic expression of the mutation
reflects the lack of an important protein, rather than
the production of a toxic one.
The above constitute client protein-driven ER stress,
however ER stress may also initiate from impaired
function of the organelle, which occurs in cells
deprived of energy sources or oxygen, or in cells
exposed to certain ER-specific toxins. We do not
understand in detail how ER stress contributes to
further organelle dysfunction and ultimately cell death.
However a framework for thinking about this has
recently emerged with the realization that unfolded
and malfolded proteins present reactive interfaces that
have not been vetted by evolution and may thus
disrupt the cellular machinery by interacting promis-
cuously and illegitimately with essential cellular
components. According to this theory, proteotoxicity
is normally held in check by the chaperones that bind
such potentially toxic protein interfaces and let go only
once the latter have been buried in the hydrophobic
cores of the properly folded client protein. ER stress
(by definition) challenges the capacity of the chaper-
ones and may permit illegitimate protein interfaces
to emerge. The ability of chaperone overexpression to
suppress ER stress signaling suggests that the need
to prevent proteotoxicity is an important driving force
in evolution of the UPR. The unifying feature of ER
stress need not be the presence of toxic moieties on the
surface of every unfolded or malfolded protein. The
common feature of diseases of protein folding might
instead be the exhaustion of a protective chaperone
reserve, which normally suppresses the potential
proteotoxicity of certain (possibly normal) folding
intermediates of ER client proteins.
The Yeast Unfolded
Protein Response
IRE1, A PROTOTYPE OF
TRANSMEMBRANE STRESS SIGNALING
Early studies on the UPR were carried out in the yeast,
Saccharomyces cerevisiae, an organism that lends itself
well to forward genetic screens. To screen for mutations
affecting the UPR, the regulatory region of yeast BiP
gene (KAR2) was fused to a reporter and mutant yeast
with suppressed activity of this reporter were sought.
The first gene thus identified was inositol requiring 1
(IRE1), so-called because its loss of function had been
previously noted to result in inositol auxotrophy. IRE1
encodes a transmembrane ER resident protein with an
N-terminal domain residing in the ER lumen and a C-
terminal domain that is exposed on the cytoplasmic
side. The membrane topology and subcellular localiz-
ation of IRE1 immediately suggest a mechanism for
transmitting information on the state of the ER
(topologically equivalent to the extracellular space) to
the cell’s interior. The lumenal domain somehow senses
ER stress, conveying the signal across the ER mem-
brane to the cytoplasmic domain, which broadcasts it
to the nucleus, turning “on” UPR target gene
expression (Figure 1).
IRE1’s C-terminal, cytoplasmic, effector-domain, is a
protein kinase and undergoes autophosphorylation
when activated by ER stress. This suggested that IRE1
might function like other transmembrane receptors that
are also protein kinases and convey their signal by
phosphorylating downstream targets, often through a
kinase relay. However, other than IRE1 itself no
substrates for the aforementioned kinase have been
identified to date.
UNCONVENTIONAL SPLICING
OF
HAC1 MRNA
The clues to understanding propagation of the UPR
signal, downstream of IRE1 again came from yeast
genetics. Mutations in two additional genes were
noted to block the yeast UPR. One of these, HAC1,
encodes a transcription factor, which binds to and
activates the promoters of the primary target genes
of the yeast UPR, the second, more mysteriously
turned out to be RLG1, which encodes for a multi-
functional enzyme involved in tRNA splicing. ER
stress promotes accumulation of HAC1 protein and
this is blocked by mutations in IRE1 and RLG1.
320
UNFOLDED PROTEIN RESPONSES
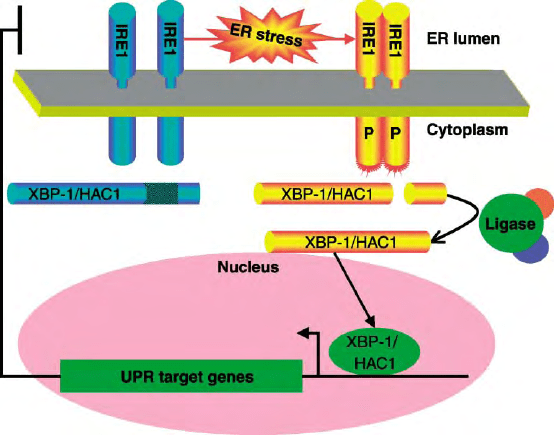
The underlying mechanisms, which turned out to be
full of surprise, were revealed in a series of
brilliant experiments carried out in the Walter and
Mori laboratories.
The regulated step in HAC1 expression entails the
removal of a 242 base internal segment at the 3
0
end
of the mature mRNA. The enzyme which precisely
cuts the mRNA at the 5
0
and 3
0
ends of this segment
turned out to be IRE1 itself, whose highly sequence-
specific endoribonucleolytic activity appears to be
subordinate to its kinase activity (though the molecu-
lar details remain to be worked out). The two ends of
the severed HAC1 mRNA are held together by
extensive base pairing and are subsequently religated
by the protein product of RLG1. IRE1 is the master
regulator of this unconventional mRNA splicing
reaction, since its kinase and endoribonuclease
are activated by ER stress. The removal of the
aforementioned internal segment of the HAC1
mRNA de-represses HAC1 translation and the protein
encoded by the processed mRNA is more stable and
transactivates its target genes more potently than that
encoded by the unprocessed mRNA.
Mutations in IRE1 and HAC1 similarly abolish most
signaling in the yeast UPR. This was convincingly
demonstrated by expression profiling which showed
that the vast majority of the genes induced by exposing
yeast to toxins that promote ER stress were no longer
activated in IRE1 or HAC1 mutant strains. Thus the
yeast UPR consists of a linear pathway in which HAC1
is an essential target of IRE1 and together the two
proteins are required for the activation of most known
UPR target genes.
Diversification in the
Metazoan UPR
CONSERVATION OF THE IRE1 PATHWAY
Systematic genomic sequencing of the metazoan
C. elegans and random cDNA sequencing from several
mammalian species, predicted proteins highly related to
yeast IRE1 in higher eukaryotes. Their common
membrane topology, localization to the ER, and
conserved kinase and endoribonuclease domains
suggested that they were indeed IRE1 homologues.
However, HAC1 homologues could not be identified by
simple sequence comparison to the yeast. Eventually,
however, a combination of forward genetic screens in
C. elegans and creative guesswork led to the identifi-
cation of XBP-1 as a functional homologue of HAC1 in
higher eukaryotes. It too encodes a transcription factor
whose expression is tightly regulated by IRE1 through
the processing of its mRNA.
Less conserved, however, is the role of the IRE1
pathway in the yeast and metazoan UPR. Whereas in
the former, IRE1 and HAC1 are absolutely required
for activating nearly all UPR target genes, in mam-
mals, IRE1 and XBP-1 are dispensible for activation of
FIGURE 1 IRE1 and unconventional splicing of its target mRNA signal the unfolded protein response. ER stress leads to oligomerization and
trans-autophosphorylation of IRE1 (indicated by the “P” on its cytoplasmic effector domain). Phosphorylation unmasks the effector function of
IRE1 (cartooned by the red bristles on the cytoplasmic domain), which consists of the endoribonucleolytic processing of its target mRNA, XBP-1 in
metazoans, and HAC1 in yeast. The two ends of the cleaved mRNA are joined together by a ligase. The unconventionally spliced XBP-1/HAC1
mRNA is more efficiently translated than the unprocessed mRNA and encodes a protein that is more stable and more effective at trans-activation of
UPR target genes in the nucleus.
UNFOLDED PROTEIN RESPONSES 321
all but a very small set of UPR targets. This is all the
more remarkable when one considers that most UPR
target genes are conserved between yeast and mammals,
it is just that the latter have found alternative means
to couple their expression to ER stress. C. elegans
occupies an intermediate position between yeast
and mammals, in that most of its identifiable UPR
targets are partially dependent on IRE1 and XBP-1,
but can to some extent also be activated independently
of that pathway.
Despite this redundancy in the mammalian UPR,
both IRE1 and XBP-1 are essential for embryonic
development (mammals have two IRE1 genes, a
nonessential beta isoform restricted in its expression
to the intestinal and bronchial epithelium and a
broadly expressed alpha isoform which is essential).
The reason(s) for the embryonic lethality of IRE1
a
and
XBP-1 null animals are not fully understood, but it is
hypothesized that in mammals the pathway may have
diverted to specify the capacity for especially high
levels of protein secretion. This hypothesis, which is
clearly in need of further experimental support,
suggests that specification of a secretory cell fate
activates a conserved stress pathway that drives a
developmental process, namely acquisition of an
apparatus required by professional secretory cells
for high-capacity protein secretion. This idea is
further supported by the observation that many
UPR target genes function far downstream in the
secretory pathway rendering it unlikely that such genes
merely relieve the stressed ER of its load of unfolded
client proteins.
ATF6 AND INTRAMEMBRANE
PROTEOLYSIS IN THE METAZOAN UPR
The identification of ATF6 by Mori and colleagues
confirmed the predicted redundancy in the metazoan
UPR. Like IRE1, ATF6 is also an ER-localized trans-
membrane protein. However, its cytoplasmic effector
domain consists of a transcription factor that activates
UPR target genes directly. In its membrane-bound form,
ATF6 is inert, as it cannot reach the nucleus. Activation
involves regulated intramembrane proteolysis, which
liberates the transcription factor part of ATF6 from the
ER membrane under conditions of ER stress (Figure 2).
The next surprise came when Brown, Goldstein, Prywes,
and colleagues identified the proteases involved in this
highly regulated event. These turned out to be the same
proteases that process and activate the membrane-
bound transcription factor, SREBP, which regulates
genes involved in sterol and fatty acid biosynthesis
and assimilation.
There are no mammalian gene knockouts reported
for ATF6 (of which at least two isoforms exist).
However, unlike cells lacking IRE1 or XBP-1, cells
lacking the proteases required for ATF6 processing are
severely impaired in UPR target gene expression. It
seems likely therefore that mammalian ATF6 has
picked up some of the role performed by IRE1 and
XBP-1/HAC1 in simpler organisms. The similarities
between ATF6 and the SREBPs highlight another
remarkable feature of the UPR. IRE1 and HAC1
mutant yeast are unable to synthesize adequate
amounts of the membrane phospholipid precursor,
inositol. Though the details remain to be worked out,
insufficiency of membrane lipid components also
signals through the yeast UPR and enzymes involved
in phospholipid metabolism are targets of IRE1 and
HAC1. In metazoans this aspect of the UPR apparently
has been split off and relegated to dedicated transcrip-
tion factors, the SREBPs that are activated by insuffi-
ciency of lipid components. However the ancient
link between the client protein-based UPR and mem-
brane lipid insufficiency signaling has remained in the
form of shared machinery for proteolytic activation of
ATF6 and SREBP.
Translational Control in the UPR
PERK COUPLES ER STRESS TO EIF2
a
PHOSPHORYLATION AND
TRANSLATIONAL REPRESSION
In addition to activated gene expression ER stress
results in a dramatic reduction in protein synthesis.
Translational repression is an active process limiting
the influx of client proteins into the stressed ER and
thus serves as a counterpart to the gene expression
program, which increases the organelle’s capacity to
process client proteins. Brostrom and colleagues and
Kaufman and colleagues noted, early on, that transla-
tional repression by ER stress is associated with
phosphorylation of the
a
-subunit of translation
initiation factor 2 (eIF2
a
) on serine 51. This phos-
phorylation site is conserved in all eukaryotes and
serves to regulate translation initiation in diverse
stressful conditions. Trimeric eIF2, in complex with
guanosine triphosphate (GTP), recruits the amino-
acylated initiator methionyl –tRNA to the small
ribosomal subunit, allowing translation initiation.
Recognition of an AUG initiation codon on the
mRNA leads to hydrolysis of GTP to GDP and
dissociation of the ribosome–eIF2 complex. To par-
ticipate in another round of translation initiation, the
GDP bound to eIF2 must be exchanged to GTP. The
enzyme catalyzing this exchange reaction (eIF2B) is
inhibited by phosphorylated eIF2.
Distinct eIF2
a
kinases were known to be activated
by distinct stress signals; PKR by double-stranded RNA
322
UNFOLDED PROTEIN RESPONSES