Hermanson G. Bioconjugate Techniques, Second Edition
Подождите немного. Документ загружается.

1000 27. Nucleic Acid and Oligonucleotide Modifi cation and Conjugation
done using fl uorescent probes to target particular regions within chromosomes. Called FISH
for fl uorescent in situ hybridization, the technique is used extensively to identify marker chro-
mosomes or chromosomal rearrangements. Since many genomic sequences are repeated, usu-
ally occurring in multiple copies within isolated regions of the chromosome, the fl uorescent
label on the DNA probe allows localization of targeted genes with high sensitivity. For a review
of FISH, see Meyne (1993).
Fluorescently labeled DNA probes also can be used in homogeneous assay systems to detect
and quantify target complementary sequences. The majority of these systems use a process of
energy transfer and fl uorescent quenching to detect hybridization phenomena. The principle of
these assays involves the labeling of two binding components that can specifi cally interact with
a target DNA. One or both of the labels may be a luminescent compound. The luminescent
quality of the fi rst label may consist of a chemiluminescent probe that can be excited through
specifi c chemical processes, producing light emission. Alternatively, the label may be a fl uores-
cent probe which can absorb light of a particular wavelength and subsequently emit light at
another wavelength.
The second label also may be a fl uorescent compound, but doesn ’t necessarily have to be.
As long as the second label can absorb the emission of the fi rst label and modulate its signal,
binding events can be observed. Thus, the two labeled DNA probes interact with each other to
produce fl uorescence modulation only after both have bound target DNA and are in enough
proximity to initiate energy transfer. Common labels utilized in such assay techniques include
the chemiluminescent probe, N -(4-aminobutyl)- N-ethylisoluminol, and reactive fl uorescent
derivatives of fl uorescein, rhodamine, and the cyanine dyes (Chapter 9). For a review of these
techniques, see Morrison (1992).
To prepare labeled DNA molecules for use in fl uorescent assays, the oligo must be fi rst deriv-
atized to contain a functional group. Any of the methods of Sections 2.1 and 2.2 (this chap-
ter) may be used to add an amine or sulfhydryl residue to specifi c regions of the DNA polymer.
Once modifi ed in this manner, the oligo may be further reacted with a fl uorescent probe to cre-
ate the fi nal derivative. However, many of the fl uorescent quenching formats exclusively specify
either 3 - or 5 -labeled DNA molecules. This is because discrete modifi cation at just one end of
the oligo assures that the label on a hybridized probe will be near enough to a second hybrid-
ized-and-labeled DNA molecule to effect the luminescent modulation necessary to make the
system viable. Multiple fl uorescent labels on nucleotides within the DNA probe would not be
affected to the same degree by a second label attached to another oligo hybridized some dis-
tance away on the target strand. Therefore, use of the terminal transferase method of adding a
modifi ed nucleoside triphosphate to the 3 end (Section 1) or 5 -phosphate modifi cation using
a carbodiimide-mediated reaction (Section 2.1) work best for creating functionalized DNA
derivatives for fl uorescent modulation techniques.
Another method of fl uorescent detection of DNA or RNA targets involves the modifi ca-
tion of a targeting oligo at both ends. In this approach, one end is modifi ed with a fl uorescent
molecule and the other end is modifi ed with another label that can be a quencher of fl uores-
cence or another fl uorescent probe able to accept energy from the fi rst label. These “molecular
beacons”, as they are called, contain terminal sequences that are able to hybridize the ends
together in solution in the absence of target. Thus, if a fl uorescent molecule is at one end of the
molecular beacon and a quencher is at the other end, in solution without a target DNA present
the fl uorescent signal would be completely quenched. In the presence of the target DNA, the
molecular beacon would hybridize to the target and open up the stem-and-loop structure of the
probe, thus forcing the fl uorophore and quencher too far away from each other to cause fl uo-
rescent quenching. The result is that specifi c target binding of the molecular beacon causes the
generation of a fl uorescent signal. The more fl uorescence that occurs the more target is present
in solution. For a review of molecular beacon technology, see Bratu (2006).
The following sections describe two methods of coupling fl uorescent labels to functional-
ized DNA probes. Other fl uorophores or quenching molecules may be attached using similar
procedures with careful reference to the properties and reactivities of such labels as discussed
in Chapter 9.
Conjugation of Amine-Reactive Fluorescent Probes to Diamine-Modifi ed DNA
DNA modifi ed with a diamine compound to contain terminal primary amines may be coupled
with amine-reactive fl uorescent labels. The most common fl uorophores used for oligonucle-
otide labeling are the cyanine dyes and derivatives of fl uorescein and rhodamine (Chapter 9).
However, any of the amine-reactive labels discussed throughout Chapter 9 are valid candidates
for DNA applications.
Some fl uorescent probes are water-insoluble and must be dissolved in an organic solvent
prior to addition to an aqueous reaction medium containing the DNA to be labeled. Even
water-soluble fl uorescent probes may be dissolved fi rst in an organic solvent to permit easy
addition to an aqueous reaction medium without hydrolysis of the reactive group. Suitable sol-
vents are identifi ed for each fl uorophore, but mainly DMF or DMSO are used to prepare a
stock solution. Some protocols utilize acetone when labeling DNA. However, avoid the used
of DMSO for sulfonyl chloride compounds as this group reacts with the solvent. For oligo-
nucleotide labeling, the amount of solvent added to the reaction mixture should not exceed
more than 20 percent (although at least one protocol calls for a 50 percent acetone addition—
Nicolas et al ., 1992).
The following protocol is a generalized method for labeling amine-modifi ed oligonucleotides
with a fl uorescent probe, such as FITC. It is based on the method of Morrison (1992).
Protocol
1. Prepare 10 nmol of a diamine-modifi ed DNA probe using the chemical methods dis-
cussed in Section 2.1 or through enzymatic derivatization using an amine-containing
nucleoside triphosphate (Section 1). Dissolve or buffer exchange the oligo into 1.0 ml of
a suitable coupling buffer for the type of amine-reactive fl uorophore utilized (see recom-
mended reaction conditions for the particular fl uorescent label in Chapter 9). For FITC,
the appropriate buffer condition for the oligo is 0.1 M sodium carbonate, pH 9.0.
2. Dissolve the fl uorophore in DMF or another suitable solvent at a concentration of
0.01 M. For FITC, this translates into a concentration of 3.89 mg/ml.
3. Add 50 l of the FITC solution to the oligo solution and mix. For the use of NHS ester
or sulfonyl chloride fl uorescent probes, add up to 150 l of the fl uorophore solution to
the DNA.
4. React overnight at room temperature.
5. Remove excess fl uorophore from the labeled oligo using gel fi ltration on a desalting resin,
dialysis, or a centrifugal concentrator.
2. Chemical Modifi cation of Nucleic Acids and Oligonucleotides 1001
1002 27. Nucleic Acid and Oligonucleotide Modifi cation and Conjugation
Conjugation of Sulfhydryl-Reactive Fluorescent Probes to Sulfhydryl-Modifi ed DNA
Fluorescent probes containing sulfhydryl-reactive groups can be coupled to DNA molecules
containing thiol modifi cation sites. The chemical derivatization methods outlined in Section 2.2
(this chapter) may be used to thiolate the oligo for subsequent modifi cation with a fl uorophore.
Appropriate fl uorescent compounds and their reaction conditions may be found in Chapter 9.
The protocol discussed in the previous section can be used as a general guide for labeling DNA
molecules.

1003
28
The study of protein interactions has become a vital research effort as a result of the
sequencing of the human genome and the genomes of other organisms. The next great challenge
beyond merely having knowledge of gene sequences is to understand the complex interplay of
the resultant protein molecules within cells as they bind, interact, and affect cellular processes.
From the many research papers that have appeared on this subject, it is becoming clear that
each protein molecule interacts with other proteins and molecules not in isolated obscurity, but
in a highly complex web of interactions, which can have far reaching effects on overall biologi-
cal function.
Protein interactions mediate virtually every cellular process. They are involved with tran-
scription, translation, transport, cell cycle control, the determination of cell type and function,
protein folding, post-translational modifi cations, signal transduction, metabolism and energy
production, cell structure and motility, the formation of biological machines, cell and organism
defense, and apoptosis. Genetic mutations or damaging modifi cations resulting in abnormal
protein interactions often are the root cause of many diseases, especially tumorigenesis.
The general types of protein–protein interactions that occur in cells include receptor–ligand,
enzyme–substrate, multimeric complex formations, structural scaffolds, and chaperones.
However, proteins interact with more targets than just other proteins. Protein interactions can
include protein–protein or protein–peptide, protein–DNA/RNA or protein–nucleic acid, protein–
glycan or protein–carbohydrate, protein–lipid or protein–membrane, and protein–small molecule
or protein–ligand. It is likely that every molecule within a cell has some kind of specifi c interaction
with a protein.
The consequences of protein interactions in effecting cellular biochemistry include the syn-
thesis, destruction, or recycling of biomolecules; the supply of energy for cellular processes;
the generation of chemical signals; activation or inhibition of proteins and enzymes; changes
in protein conformation and structure; the creation of new binding sites or active centers in
proteins; the motility of cells, tissues, and organisms; transport of molecules within cells or
into/out of cells; and the formation of cellular structures and compartments.
Protein interactions can be described in relative ways related to the strength of the binding
that takes place between the two molecules and the time that the interaction lasts. Any inter-
acting molecules can be characterized as having either (1) high affi nity (strong) or low affi nity
(weak) binding and (2) stable (long lasting) or transient (short-lived) binding. Thus, a given
Bioconjugation in the Study of Protein Interactions
protein interaction may be described in one of four possible ways: strong and stable, strong
and transient, weak and stable, or weak and transient. Interactions of the weak and stable type
may at fi rst seem like an oxymoron, but often multiple weak interactions can take place simul-
taneously with a target and the combined “avidity” makes the resultant complex stable.
Of the large number of protein interactions that take place in cells, perhaps the vast major-
ity may be described as transient. Most proteins that modify other molecules do so very rapidly
and so interact only briefl y with their substrates or binding partners (i.e., enzymes). In addition,
since proteins within cells are highly compartmentalized, the affi nity of most interactions doesn ’t
have to be very great, because each potential binding partner is within short diffusion distances
and the relative concentration of molecules within these small volumes is high.
Of course, the designations of strong, weak, stable, or transient are all subjective terms. They
mainly result from the outcome of affi nity capture experiments of binding partners on insolu-
ble supports or the analytical determination of the kinetic parameters of specifi c binding inter-
actions. In general, if the affi nity constant of a protein interaction is strong enough to allow
a binding partner to be captured and purifi ed using an immobilized affi nity ligand, then the
interaction can be described as being reasonably strong. This usually correlates to an affi nity
constant of 10
6
/M. Conversely, interactions having affi nity constants of 10
6
/M are often too
weak to survive the washing steps needed to isolate the interacting protein on a solid phase
affi nity support. Quantitative measurement of the affi nity constant between interacting proteins
and the half-life of the interaction may be done using surface plasmon resonance (SPR) tech-
niques (Homola et al., 1995; Homola et al., 1999).
The vast network of protein–protein interactions that have been deciphered in recent years
has grown to include literally thousands of proteins in a sometimes-chaotic dance of complex-
ity. Using the two-hybrid method for instance, which involves the use of split transcription fac-
tor fusion proteins that allow detection of interacting proteins by activation of reporter gene
expression, many putative protein–protein interaction partners have been identifi ed in yeast
(Fields and Song, 1989; Chien et al., 1991; Criekinge and Beyaert, 1999). A map of these inter-
actions looks a lot like a picture of the myriad nodes of addresses on a vast network of com-
puters, such as the interconnections that make up the Internet (Jeong et al., 2001). Although
most of the proteins in an organism have only a few partners that interact with them, major
hubs in protein “interactomes” can have up to 10–20 links with other proteins, indicating that
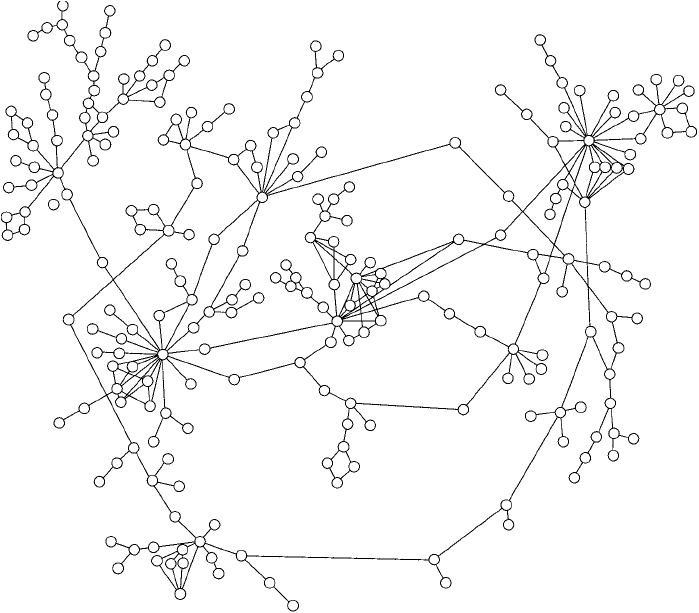
these key proteins are critical players in cell vitality Figure 28.1 .
Through the growing knowledge of protein–protein interactions and their corresponding gene
sequences major interaction domains on protein surfaces are being identifi ed. These relatively
conserved amino acid sequences create structural motifs that are designed to bind with certain
targeted sequences in other proteins or molecules (Pawson and Nash, 2003; Ingham et al., 2005).
Using this information, many common binding regions on proteins can be identifi ed just through
their gene sequences.
However, it is much more diffi cult to characterize the interactions of proteins with no
known interaction domains. The traditional “lock and key ” approach to conceptualizing
binding pairs is far too simplistic to allow easy visual identifi cation of interacting surfaces on
the complex three-dimensional space making up the topography of protein molecules. Even
in those instances where protein interacting partners have been crystallized together and their
three-dimensional structures determined, it ’s obvious from the molecular models that it would
be diffi cult or impossible to identify visually the site of interaction without having such struc-
tural data in place beforehand. For this reason, experimental schemes are needed that are more
1004 28. Bioconjugation in the Study of Protein Interactions
elaborate than just knowledge of genetic sequences or interaction domains to characterize the
majority of specifi c interactions proteins undergo. Even when two-hybrid studies indicate the
probability of a protein interaction occurring, it still doesn ’t provide information on the nature
of the interaction, the binding sites, or its function.
The techniques developed to study protein interactions can be divided into a number of major
categories (Table 31.1), including bioconjugation, protein interaction mapping, affi nity capture,
two-hybrid techniques, protein probing, and instrumental analysis (i.e., NMR, crystallography,
mass spectrometry, and surface plasmon resonance). Many of these methods are dependent on the
use of an initial bioconjugation step to discern key information on protein interaction partners.
Many of the methods developed to study protein interactions use the bait/prey model to
detect interacting partners (Phizicky and Fields, 1995; Archakov et al., 2003 Piehler, 2005).
The bait protein is a purifi ed protein (often recombinant) that is used to lure and capture a
putative interacting protein or biomolecule. The bait protein may be immobilized to a solid
phase for affi nity separations or be used in solution. It also may be fusion tagged (i.e., GST or
6 His) or labeled with a detectable molecule, such as a fl uorescent probe. It often is the case
Figure 28.1 A small segment of the yeast interactome. The spheres represent proteins and the interconnecting
lines are identifi ed protein interactions. Many proteins are seen to interact with one or two other proteins, but
some can have over a dozen other interacting partners.
28. Bioconjugation in the Study of Protein Interactions 1005
1006 28. Bioconjugation in the Study of Protein Interactions
that there exists an antibody specifi c for the bait protein to use for detection or in recovery of
the interacting complexes.
The prey is a protein, protein complex, or other biomolecule that interacts with the bait
protein. It can be captured by its specifi c affi nity interaction with the bait. Since many pro-
tein interactions may involve low affi nity binding events or are transient, the use of chemical
crosslinking techniques can greatly facilitate analysis of an interacting prey protein. In fact, the
use of bioconjugation to “fi x ” interacting proteins is a powerful route to capturing weak affi n-
ity or transiently interacting molecules, as the crosslinked complexes can be isolated and ana-
lyzed without loss of some components.
The following sections describe the use of bioconjugation reagents for the study of protein
interactions. These reagents and techniques can be used to crosslink and capture interacting pro-
teins, to investigate the binding sites involved with interactions, and to identify which peptide
regions or amino acids participate in the binding event. In addition to the bioconjugation rea-
gents described in this section, the reader also is referred to Chapter 16, entitled Mass Tags and
Isotope Tags, wherein some of those reagents also can be used to investigate interacting proteins.
1. Homobifunctional Crosslinking Agents
One of the fi rst applications of bioconjugate techniques was the use of simple homobifunctional
compounds (see Chapter 4) to capture interacting proteins in biological samples. As the name
of this type of reagent implies, these compounds have reactive groups on each end of a spacer
arm that are identical and react with the same type of functionality on different proteins. For
instance, early development of crosslinking compounds resulted in the creation of homobifunc-
tional amine-reactive reagents using either imidoesters or NHS esters as the reactive groups.
Both of these reagent types could be reacted with a complex protein sample mixture to result in
effi cient conjugation of proteins through their available amine groups. The reactive groups on
these compounds survive long enough in a physiological pH environment to effectively capture
and covalently link proteins in proximity to one another. In this way, any proteins undergoing
specifi c biological interactions can be “fi xed ” by chemical conjugation and thus link together
permanently interacting complexes for analysis.
However, as one might expect, this strategy is more or less a crude “shotgun” approach,
wherein every protein in the sample has the potential to be crosslinked with other proteins
regardless of whether they are undergoing specifi c interactions or not. The use of homobifunc-
tional reagents to study protein interactions therefore may result in high noise levels or many
false positives, because of their non-selective crosslinking characteristics. The end result often
makes it diffi cult to identify conjugated proteins that are specifi cally interacting from those pro-
teins crosslinked just due to random collisions.
However, even acknowledging their disadvantages, homobifunctional crosslinkers have
been used successfully to investigate many protein–protein interactions, within cells and within
lysates or protein solutions. The key to capturing true interacting proteins while limiting the
degree of nonspecifi c conjugation is to optimize the amount of crosslinker at the lowest pos-
sible concentration. Using too high a concentration will extensively conjugate all proteins, even
those that are not specifi cally interacting at the moment. Using too low a concentration will not
conjugate effectively even interacting proteins. Therefore, optimization typically is needed to
determine the best concentration of homobifunctional crosslinker for a particular application.
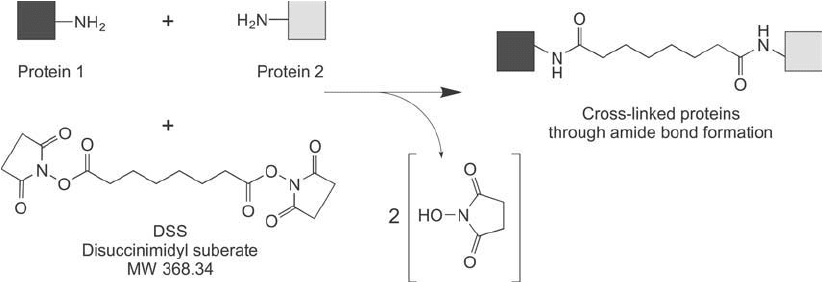
1.1. DSS and BS
3
Two crosslinking agents that have been used extensively to study protein interactions are disuc-
cinimidyl suberate (DSS) and bis-sulfosuccinimidyl suberate (BS
3
) (see Chapter 4, Section 1.2).
Both reagents contain an 8-carbon spacer arm built from suberate core and have reactive esters at
each end that couple with amines to form amide bonds. They differ, however, in the fact that DSS
contains NHS esters and BS
3
contains negatively charged sulfo-NHS esters. BS
3
therefore is water-
soluble and will not penetrate cell membranes, whereas DSS is hydrophobic, water-insoluble, and
is membrane permeable. For this reason, BS
3
can be used to study cell-surface–protein interactions
(Friedrichson and Kurzchalia, 1998; Simons et al., 1999), while DSS can be used to study intra-
cellular protein interactions (Ishmael et al., 2005). The reaction of DSS to capture and crosslink
interacting proteins is shown in Figure 28.2 .
The following protocol represents a generalized strategy for crosslinking interacting proteins
using either DSS or BS
3
. The buffer conditions and reagent amounts are gleamed from published
procedures, but the exact quantities should be optimized for each protein interaction studied.
Protocol
1. Suspend cells at 25 10
6
cells/ml in PBS (pH 8.0).
2. Wash cells 3 times with ice-cold PBS (pH 8.0) to remove amine-containing culture media
and extracellular proteins from the cells.
3. For cell-surface interaction studies, add ligands to the cells and incubate for 1 hour at
4°C.
4. Dissolve DSS or BS
3
in dry DMSO at a concentration of 25 mM. Note: BS
3
may be added
directly to PBS buffer or dissolved as a stock solution in DMSO.
5. Add an aliquot of the DSS or BS
3
solution to the reaction medium to obtain a fi nal con-
centration of 0.5–5 mM. Note: Simons et al. (1999) successfully used a concentration of
0.5 mM BS
3
with Madin-Darby canine kidney (MDCK) cells permanently expressing a
GPI-anchored form of growth hormone decay accelerating factor (GH-DAF) to crosslink
the protein interaction complexes on the cell surfaces.
Figure 28.2 DSS can capture protein interacting partners through amide bond crosslinks.
1. Homobifunctional Crosslinking Agents 1007
1008 28. Bioconjugation in the Study of Protein Interactions
6. Incubate the reaction mixture for 30 minutes at room temperature. To reduce active
internalization of BS
3
into cells, this incubation may be performed at 4°C.
7. Quench the reaction by adding an aliquot of 1 M Tris, pH 7.5, to give a fi nal concentra-
tion of 10–20 mM.
8. Incubate the quenching reaction for 15 minutes at room temperature.
9. Lyse cells and analyze the protein interactions by electrophoresis, Western blotting, and
mass spec.
1.2. Heavy Atom, Deuterated Crosslinking Agents
Another approach to the study of protein interactions using homobifunctional crosslinkers uses
the incorporation of isotopes, like deuterium, into the reagent structure to produce a heavy
atom analog suitable for mass spec analysis. In this technique, the normal or light H (hydro-
gen) version of a crosslinker is used in an equal molar ratio to a heavy D (deuterium) version
to capture interacting proteins through covalent conjugation. The heavy and light analogs are
reacted with a sample at the same time, so that each form of the reagent will have an equal
chance of reacting with an interaction complex. Subsequent proteolytic digestion of the sample
(i.e., with trypsin) creates peptide fragments, some of which will contain covalent crosslinks
from the heavy or light crosslinkers. In addition, these crosslinked peptides will have an equal
chance of having either a light atom linker or a heavy atom linker holding them together.
Mass spec analysis of the peptide fragments formed by this process yields pairs of MS peaks
differing only by the mass change caused by the substitution of deuterium atoms for hydrogen
atoms in half of the crosslinks. Thus, searching for MS peaks in the data that differ by the
number of deuterium substitutions immediately will identify peptides from the interacting pro-
teins that have been captured by the crosslinking process.
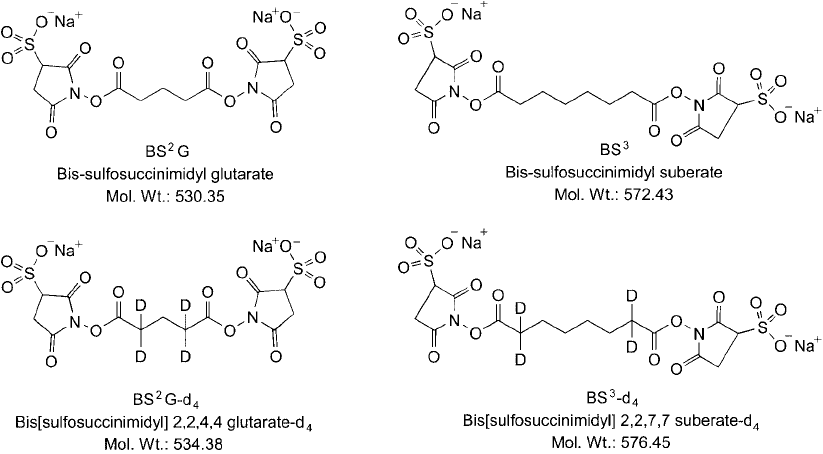
Using this approach, homobifunctional crosslinking agents containing sulfo-NHS esters have
been developed based on the core structures of glutaric and suberic acids. The suberate-based
crosslinker also is known as BS
3
and has been described previously (Section 1.1, this chapter).
These two standard amine-reactive reagents then are modifi ed at two carbons of their respec-
tive cross-bridge structures to contain two pairs of deuterium atoms, increasing their molecular
mass by exactly 4 from the normal hydrogen atom analogs.
These heavy atom reagent pairs are termed BS
2
G-d
4
(bis[sulfosuccinimidyl] 2,2,4,4
glutarate-d
4
) and BS
3
-d
4
(bis[sulfosuccinimidyl] 2,2,7,7 suberate-d
4
), and their light atom ana-
logs are called BS
2
G-d
0
(bis[sulfosuccinimidyl] glutarate-d
0
) and BS
3
-d
0
(bis[sulfosuccinimidyl]
suberate-d
0
) (Thermo Fisher) ( Figure 28.3 ). The sulfo-NHS esters of these four compounds all
react equally well with amine groups on proteins to form amide bonds. Therefore, there are
virtually no differences in properties or reactivities between the heavy and light versions of
these reagents.
The conjugation of interacting proteins with heavy/light crosslinkers potentially can result
in a number of derivatives having unique atomic mass units (amu) observed by mass spec. For
instance, any of these crosslinkers might react with amines on interacting proteins at both ends
to form amide bonds. Alternatively, one end of these homobifunctional compounds may react
with a protein to form an amide bond while the other end hydrolyzes, resulting in loss of the
sulfo-NHS group to form a carboxylate. Thus, the heavy/light crosslinking pairs may have two
potential amu results when peptides are analyzed by mass spec. Table 31.2 shows the potential
amu contributions for each of the possible products formed by the reactions of the heavy/light
crosslinkers with proteins.
This technique has been described as a general method of studying protein–protein interac-
tions as well as a method for investigating the three-dimensional structure of individual proteins
(Muller et al., 2001; Back et al., 2003; Dihazi and Sinz, 2003; Sinz, 2003; Sinz, 2006). It also has
been used for the study of the interactions of cytochrome C and ribonuclease A (Pearson et al.,
2002), to investigate the interaction of calmodulin with a specifi c peptide binder (Kalkhof et al.,
2005a; Schmidt et al., 2005), and for probing laminin self-interaction (Kalkhof et al., 2005b).
In practice, sample concentrations typically are kept in the micromolar range to limit the
widespread conjugation of molecules not specifi cally interacting or to limit intermolecular con-
jugation when studying the structure of a single protein. Therefore, most protein concentra-
tions will be in the microgram/ml range prior to the addition of crosslinkers. Even in cases
wherein a single purifi ed protein is crosslinked to study its three-dimensional structure, limit-
ing the amount of crosslinker in the reaction mixture will help to limit polymerization of the
protein. However, successful results have been obtained by this method using up to a 200-fold
excess of crosslinker over the concentration of protein in solution.
The following protocol is based on the published methods as well as the specifi c instructions
provided with the heavy atom crosslinkers from Thermo Fisher Scientifi c. For new applications,
the amount of crosslinkers added to the sample will have to be optimized to obtain useful
information about the interactions.
Figure 28.3 The homobifunctional crosslinkers BS
2
G and BS
3
can be used to capture protein interactions
through amide bond formation. The deuterium-labeled analogs of these reagents can be used to differentiate
samples by mass spec.
1. Homobifunctional Crosslinking Agents 1009