Bhushan B. Nanotribology and Nanomechanics: An Introduction
Подождите немного. Документ загружается.

13 Computer Simulations of Nanometer-Scale Indentation and Friction 657
13.2 Computational Details
Molecular dynamics simulations are straightforward to describe: given a set of ini-
tial conditions and a way of mathematically modeling interatomic forces, Newton’s
(or equivalent) classical equation of motion is numerically integrated [45]
F = ma , (13.1a)
−∇E = m(∂
2
r/∂t
2
) , (13.1b)
where F is the force oneach atom, m is the atomicmass, a is the atomicacceleration,
E is the potential energy felt by each atom, r is the atomic position, and t is time.
The forces acting on any given atom are calculated, and then the atoms move a short
increment ∂t (called a time step) forward in time in response to these applied forces.
This is accompanied by a change in atomic positions, velocities and accelerations.
The process is then repeated for some specified number of time steps.
The output of these simulations includes new atomic positions, velocities, and
forces that allow additional quantities such as temperature and pressure to be deter-
mined. As the size of the system increases, it is useful to render the atomic positions
in animated movies that reveal the responses of the system in a qualitative manner.
Quantitative data can be obtained by analyzing the numerical output directly.
The following sections review the way in which energies and forces are calcu-
lated in MD simulations and the important approximations that are used to realis-
tically model the friction that occurs in experiments with smaller systems of only
a few tens of thousands of atoms in simulations. The reader is referred to additional
sources [46–52] for a more comprehensive overview of MD simulations (includ-
ing computer algorithms) and the potentials that are used to calculate energies and
forces in MD simulations.
13.2.1 Energies and Forces
There are several different approaches by which interatomic energies and forces are
determined in MD simulations. The most theoretically rigorous methods are those
that are classified as ab initio or first principles. These techniques, which include
density functional theory [53,54] and quantum chemical ab initio [55] methods, are
derived from quantum mechanical principles and are generally both the most accu-
rate and the most computationally intensive. They are therefore limited to a small
number of atoms (< 500), which has limited their use in the study of friction. Alter-
natively, empirical methods are functions containingparameters that are determined
by fitting to experimental data or the results of ab initio calculations [50]. These
techniques can usually be relied on to correctly describe qualitative trends and are
often the only choice available for modeling systems containing tens of thousands,
millions, or billions of atoms. Empirical methods have therefore been widely used
in studies of friction. Semi-empirical methods, including tight-binding methods, in-
clude some elements of both empiricalmethods and ab initio methods.For instance,
658 Susan B. Sinnott et al.
they require quantum mechanical information in the form of, for example, on-site
and hopping matrix elements, and include fits to experimental data [56].
Empirical methods simplify the modeling of materials by treating the atoms
as spheres that interact with each other via repulsive and attractive terms that can
be either pairwise additive or many-body in nature. In this approach, electrons are
not treated explicitly, although it is understood that the interatomic interactions are
ultimately dependent on them. As discussed in this section, some empirical meth-
ods explicitly include charge through classical electrostatic interactions, although
most methods assume charge-neutral systems. The repulsive and attractive func-
tional forms generally depend on interatomic distances and/or angles and contain
adjustable parameters that are fit to ab initio results and/or experimental data.
The main strength of empirical potentials is their computational speed. Recent
simulations with these approaches have modeled billions of atoms [57], something
that is not possible with ab initio or semi-empirical approaches at this time. The
main weakness of empirical potentials is their lack of quantitative accuracy, espe-
cially if they are poorly formulated or applied to systems that are too far removed
from the fitting database used in their construction. Furthermore, because of the dif-
ferences in the nature of chemical bonding in various materials, such as covalent
bonding in carbon versus metallic bonding in gold, empirical methods have been
historically derived for particular classes of materials. They are therefore generally
nontransferable,although some methods havebeen shown to be theoretically equiv-
alent [51,58], and in recent years there has been progress towards the development
of empirical methods that can model heterogeneous material systems [59–64].
Several of the most important and common general classes of empirical meth-
ods used for calculating interatomic energies and forces in materials, the so-called
potentials, are reviewed here. The first to be considered are the potentials that are
used to model covalently bound materials, including the bond-order potential and
the Stillinger–Weber potential.
The bond-order potential was first formulated by Abell [65] and subsequently
developedand parameterizedby Tersoff forsilicon and germanium[66,67],Brenner
and coworkers for hydrocarbons [52,68,69], Dyson and Smith for carbon–silicon–
hydrogen systems [70], Sinnott and coworkers for carbon–oxygen–hydrogen sys-
tems [71], and Graves and coworkers [72] and Sinnott and coworkers [73] for fluo-
rocarbons.
The bond-order potential has the general functional form
E =
i
j(>i)
[V
R
(r
ij
)−b
ij
V
A
(r
ij
)] (13.2)
where V
R
(r)andV
A
(r) are pair-additive interactions that model the interatomic re-
pulsion and electron–nuclear attraction, respectively. The quantity r
ij
is the distance
between pairs of nearest-neighbor atoms i and j,andb
ij
is a bond-order term that
takes into account the many-body interactions between atoms i and j, including
those due to nearest neighbors and angle effects. The potential is short-ranged and
only considers nearest neighbor bonds. To model long-range nonbonded interac-
13 Computer Simulations of Nanometer-Scale Indentation and Friction 659
tions, the bond-order potential is combined with pair-wise potentials either directly
through splines [74] or indirectly with more sophisticated functions [75].
The Stillinger–Weber potential [76] potential was formulated to model silicon,
with a particular emphasis on the liquid phases of silicon. It includes many-body
interactions in the form of a sum of two- and three-body interactions
E =
ij
V
2
ij
(r
ij
)+
jik
V
3
jik
(r
ij
,r
ik
) , (13.3)
where V
2
is a pair-additive interaction and V
3
is a three-body term. The three-body
term includes an angular interaction that minimizes the potential energy for tetrahe-
dral angles. This term favors the formation of open structures, such as the diamond
cubic crystal structure.
The second potential is the embedded atom method (EAM) approach [77,78]
and related methods [79], which were initially developed for modeling metals and
alloys. The functional form in the EAM is
E =
i
F(ρ
i
)+
i> j
Φ(r
ij
) , (13.4)
where F is called the embedding energy. This term models the energy due to em-
bedding an atom into a uniform electron gas with a uniform compensating posi-
tive background (jellium) of density ρ
i
that is equal to the actual electron density
of the system. The term Φ(r
ij
) is a pairwise functional form that corrects for the
jellium approximation. Several parameterizations of the EAM exist (see, for exam-
ple, [77,78,80–82]) and it has recently been extendedto model nonmetallic systems.
For example, the modified EAM (MEAM) approach [64,83,84] was developed so
that EAMcouldbe appliedto metal oxides[60] andcovalentlyboundmaterials [84].
The third method is the general class of Coulomb or multipole interaction po-
tentials used to model charged ionic materials or molecules [85]. In this formalism,
an energy term is given as
E =
i
j(>i)
[(q(r
i
)q(r
j
)/r
ij
)] , (13.5)
where q(r
i
) is the charge on atom i and r
ij
is the distance between atoms i and j.
More complex formalisms that take into account, say, the Madelung constant in
the case of ionic crystals, are used in practice. In general, the charges are held
fixed, but methods that allow charge to vary in a realistic manner have been de-
veloped [61,86].
Lastly, long-range van der Waals or related forces are typically modeled with
pairwise additive potentials. A widely used approximation is the Lennard–Jones
(LJ) potential [87], which has the following functional form:
E = 4ε
i
j(>i)
(σ/r
ij
)
12
−(σ/r
ij
)
6
. (13.6)
In this approachε and σ are parametersand r
ij
is the distance between atoms i and j.
660 Susan B. Sinnott et al.
All of thesepotentialsare widely usedin MD simulationsof materials, including
studies of friction, lubrication, and wear.
13.2.2 Important Approximations
Several approximations are typically used in MD simulations of friction. The first
is the use of periodic boundary conditions (PBCs) and the minimum image con-
vention for interatomic interactions [48]. In both cases the simulation supercell is
surrounded by replicas of itself so that atoms (or phonons, etc.) that exit one side
of the supercell remerge into the simulation through the opposite side of the super-
cell. In the minimum image convention an atom interacts either with another atom
in the supercell or its equivalent atom in a surrounding cell depending on which
distance to the atom is shortest. This process is illustrated in Fig. 13.1. In this con-
vention supercells must be large enough that atoms do not interact with themselves
over the periodic boundaries. In computational studies of friction and wear, PBCs
are usually applied in the two dimensions within the plane(s) of the sliding sur-
face(s). The strength of this approach is that it allows a finite number of atoms to
model an infinite system. However, the influence of boundaries on system dynam-
ics is not completely eliminated; for example, phonon scattering due to the periodic
boundaries can influence heat transport and therefore frictional properties of sliding
interfaces.
Another important tool that is often used in MD simulations of friction is ther-
mostats to regulate system temperature. In macroscopic systems, heat that is gener-
ated from friction is dissipated rapidly from the surface to the bulk phonon modes.
Because atomistic computer simulations are limited systems that are many orders
of magnitude smaller than systems that are generally studied experimentally, ther-
mostats are needed to prevent the system temperature from rising in a nonphysical
manner. Typically in simulations of indentation or friction, the thermostat is applied
X
Fig. 13.1. Illustration of pe-
riodic boundary conditions
consisting of a central simu-
lation cell surrounded by
replica systems. The solid
arrows indicate an atom
leaving the central box and
re-entering on the opposite
side. The dotted arrows il-
lustrate the minimum image
convention

13 Computer Simulations of Nanometer-Scale Indentation and Friction 661
to a region of the simulation cell that is well removed from the interface where
friction and indentation is taking place. In this way, local heating of the interface
that occurs as work is done on the system, but excess heat is efficiently dissipated
from the system as a whole. In this manner the adjustment of atomic temperatures
occurs away from the processes of interest, and simplified approximations for the
friction term can be used without unduly influencing the dynamics produced by the
interatomic forces.
There are several different formalisms for atomistic thermostats. The simplest
of these controls the temperature by intermittently rescaling the atomic velocities to
values corresponding to the desired temperature [88] such that
v
new
v
old
2
=
T
T
ins
, (13.7)
where v
new
is the rescaled velocity, and v
old
is the velocity before the rescaling. This
approach, which is called the velocity rescaling method, is both simple to imple-
ment and effective at maintaining a given temperature over the course of an MD
simulation. It was consequently widely used in early MD simulations. The velocity
rescaling approach does have some significant disadvantages, however. First, there
is little theoretical basis for the adjustment of atomic velocities, and the system
dynamics are not time-reversible, which is inconsistent with classical mechanics.
Second, the rate and mode of heat dissipation are disconnected from system proper-
ties, which may affect system dynamics. Lastly, for typical MD simulation system
sizes, the averaged quantities that are obtained, such as pressure for instance, do not
correspond to values in any thermodynamic ensemble.
For these reasons, more sophisticated methods for maintaining system temp-
eratures in MD simulations have been developed. The Langevin dynamics ap-
proach [48], which was originally developed from the theory of Brownian motion,
falls into this category. In this approach, terms are added to the interatomic forces
that correspond to a random force and a frictionalterm [46,89,90]. Therefore,New-
ton’s equation of motion for atoms subjected to Langevin thermostats is given by
the following equation rather than (13.1a, 13.1b):
ma = F−mξv+ R(t) , (13.8)
where F are the forces due to the interatomic potential, ξ is a friction coefficient,
m and v are the particle’s mass and velocity, respectively, and R(t) is a random
force that acts as “white noise”. The friction term can be formulated in terms of
a memory kernal, typically for harmonic solids [91–93], or a friction coefficient can
be approximated using the Debye frequency. The random force can be given by
a Gaussian distribution where the width, which is chosen to satisfy the fluctuation-
dissipation theorem, is determined from the equation
R(0)R(t) = 2mk
B
Tξδ(t) . (13.9)
Here, the function R is the random force in (13.8), m is the particle mass, T is
the desired temperature, k
B
is Boltzmann’s constant, t is time, and ξ is the friction

662 Susan B. Sinnott et al.
coefficient. It should be noted that the random forces are uncoupled from those
at previous steps, which is denoted by the delta function. Additionally, the width
of the Gaussian distribution from which the random force is obtained varies with
temperature. Thus, the Langevin approach does not require any feedback from the
current temperature of the system as the random forces are determined solely from
(13.9).
In the early 1980s, Nosé developed a new thermostat that corresponds directly
to a canonical ensemble (system with constant temperature, volume and number of
atoms) [94,95], which is a significant advance from the methods described so far. In
this approach, Nosé introduces a degree of freedom s that corresponds to the heat
bath and acts as a time scaling factor, and adds a parameter Q that may be regarded
as the heat bath “mass”. A simplified form of Nosé’s method was subsequently im-
plemented by Hoover [46] that eliminated the time scaling factor whilst introducing
a thermodynamic friction coefficient ζ. Hoover’s formulation of Nosé’s method is
therefore easy to use and is commonly referred to as the Nosé–Hoover thermostat.
When this thermostat is applied to a system containing N atoms, the equations
of motion are written as (dots denote time derivatives):
˙r
i
=
p
i
m
i
, ˙p
i
= F
i
−ζp
i
,
˙
ζ =
1
Q
⎛
⎜
⎜
⎜
⎜
⎜
⎜
⎝
N
i=1
p
2
i
m
i
−N
f
k
B
T
⎞
⎟
⎟
⎟
⎟
⎟
⎟
⎠
, (13.10)
where r
i
is the position of atom i, p
i
is the momentum and F
i
is the force applied to
each atom. The last equation in (13.10) containsthe temperaturecontrol mechanism
in the Nosé–Hoover thermostat. In particular, the term between the parentheses on
the right-hand side of this equation is the difference between the system’s instanta-
neous kinetic energy and the kinetic energy at the desired temperature.If the instan-
taneous value is higher than the desired one, the friction force will increase to lower
it and vice versa.
It should be pointed out that the choice of the heat bath “mass” Q is arbitrary
but crucial to the successful performance of the thermostat. For example, a small
value of Q leads to rapid temperature fluctuation while large Q values result in inef-
ficient sampling of phase space. Nosé recommended that Q should be proportional
to N
f
k
B
T and should allow the added degree of freedom s to oscillate around its av-
eraged value at a frequency of the same order as the characteristic frequency of the
physical system [94,95]. If ergodic dynamic behavior is assumed, the Nosé–Hoover
thermostat will maintain a well-defined canonical distribution in both momentum
and coordinate space. However,for small systems where the dynamic is not ergodic,
the Nosé–Hoover thermostat fails to generate a canonical distribution. Therefore,
more sophisticatedalgorithms based on the Nosé–Hooverthermostathavebeen pro-
posed to fix its ergodicity problem; for example, the “Nosé–Hoover chain” method
of Martyna et al. [96]. However, these complex thermostats are not as easy to ap-
ply as the Nosé–Hoover thermostat due to the difficult evaluation of the coupling
parameters for each different case and the significant computationalcost [97]. From
a practical pointof view, if the molecularsystem is largeenough that the movements
13 Computer Simulations of Nanometer-Scale Indentation and Friction 663
of the atoms are sufficiently chaotic, ergodicity is guaranteed and the performance
of the Nosé–Hoover thermostat is satisfactory [25].
In an alternative approach, Schall et al. recently introduced a hybrid continuum-
atomistic thermostat[98]. In thismethod,an MD system is dividedinto grid regions,
and the average kinetic energy in the atomistic simulation is used to define a tem-
perature for each region. A continuum heat transfer equation is then solved stepwise
on the grid using a finite difference approximation, and the velocities of the atoms
in each grid region are scaled to match the solution of the continuum equation. To
help account for a time lag in the transfer of kinetic to potential energy, Hoover
constraining forces are added to those from the interatomic potential. This process
is continued, leading to an ad hoc feedback between the continuum and atomistic
simulations. The main advantage of this approach is that the experimental thermal
diffusivitycan beused in thecontinuumexpression,leadingto heattransfer behavior
that matches experimental data. For example, in metals the majority of the thermal
properties at room temperature arise from electronic degrees of freedom that are
neglected with strictly classical potentials. This thermostat is relatively straightfor-
ward to implement, and requires only the interatomic potential and the bulk thermal
diffusivity as input. It is also appropriate for nonequilibrium heat transfer, such as
occurs as heat is dissipated from sliding surfaces moving at high relative velocities.
Cushman et al. [99, 100] developed a unique alternative to the grand canon-
ical ensemble by performing a series of grand canonical Monte Carlo simulat-
ions [48,101] at various points along a hypothetical sliding trajectory. The results
from these simulations are then used to calculate the correct particle numbers at
a fixed chemical potential, which are then used as inputs to nonsliding, constant-
NVE MD simulations at each of the chosen trajectory points. The sliding speed can
be assumed to be infinitely slow because the system is fully equilibrated at each step
along the sliding trajectory. This approach offers a useful alternative to continuous
MD simulations that are restricted to sliding speeds that are orders of magnitude
larger than most experimental studies (about 1m/s or greater).
To summarize, this section provides a brief review and description of compo-
nents that are used in atomistic, molecular dynamics simulation of many of the pro-
cesses related to friction, such as indentation, sliding, and wear. The components
discussed here include the potential energy expression used to calculate energies
and forces in the simulations, periodic boundary conditions and thermostats. Each
of these components has their own strengths and weaknesses that should be well-
understood both prior to their use and in the interpretation of results. For example,
general principles related to liquid lubrication in confined areas may be most easily
understood and generalized from simulations that use pair potentials and may not
require a thermostat. On the other hand, if one wants to study the wear or indenta-
tion of a surface of a particular metal, then EAM or other semiempirical potentials,
together with a thermostat, would be expected to yield more reliable results. If one
requires information on electronic effects, ab initio or semi-empirical approaches
that include the evaluation of electronic degrees of freedom must be used. Thus, the
best combination of components for a particular study depends on the chemical na-
664 Susan B. Sinnott et al.
ture of the system of interest, the processes being simulated, the type of information
desired, and the available computational resources.
13.3 Indentation
It is critical to understand the nanometer-scale properties of materials that are be-
ing considered for use as new coatings with specific friction and wear behavior.
Experimental determination of these properties is most frequently done with the
AFM, which provides a variety of data related to the interaction of the microscope
tips with the sample surface [102–104]. In AFM experiments,the tip has a radius of
about 1–100 nm and is pressed againstthe surfaceunder ambient conditions(in air),
ultrahigh vacuum (UHV) conditions, or in a liquid. The microscope tip can either
move in the direction normal to the surface, which is the case in nanoindentation
studies, or raster across the surface, which is the case in surface imaging or friction
studies. Sliding rates of 1nm/s–1 µ/s are typically used, which are many orders of
magnitude slower than the rates used in MD simulations of sliding or indentation
of around 1–100m/s. As discussed in the previous section, the higher rates used
in computational simulations are a consequence of modeling full atomic motion,
which occurs on a femto- to picosecond timescale, and the stepwise solution of the
classical equations of motion, which makes the large number of simulation steps
needed to reach experimental timescales computationally impossible with current
processor speeds.
As the tip moves either normal to or across the surface, the forces acting upon it
as a result of its interactions with the surface are measured. When the tip is moved
in the surface normal direction, it can penetrate the surface on the nanometer scale
and provide information on the nanometer-scale mechanical properties of the sur-
face [105,106]. The indentation process also causes the force on the tip to increase,
and the rate of increase is related to both the depth of indentation and the properties
of the surface. The region of the force curve that reflects this high force is known as
the repulsive wall region [102], or, when considered without any lateral motion of
the tip, an indentation curve. When the tip is retracted after indentation, enhanced
adhesion between the tip and surface relative to the initial contact can result. This
phenomenon is indicated by hysteresis in the force curve.
Tip–surface adhesion can result from the formation of chemical bonds between
the tip and the sample, or from the formation of liquid capillaries between the mi-
croscope tip and the surface caused by the interaction of the tip with a layer of liquid
contamination on the surface. The latter case is especially prevalent in AFM studies
conducted in ambient environments. In the case of clean metallic systems, the sam-
ple can wet the tip or the tip can wet the sample in the form of a connective “neck”
of metal atoms between the surface and the tip that can lead to adhesion. In the case
of polymeric or molecular systems, entanglementof molecules that are anchored on
the tip with molecules anchored on the sample can be responsible for force curve
hysteresis.
13 Computer Simulations of Nanometer-Scale Indentation and Friction 665
In the case of horizontal rastering of AFM tips across surfaces, the force curve
data provide a map of the surface that is indicative of the surface topography [107].
If the deflection of the tip in the lateral direction is recorded while the tip is being
rastered, a friction map of the surface [20] is produced.
The restof this section discusses someof the importantinsightsand findingsthat
have been obtained from MD simulations of nanoindentation. These studies have
not only provided insight into the physical phenomena responsible for the qualita-
tive shapes of AFM force curves, they have also revealed a wealth of atomic-scale
phenomena that occur during nanoindentation that was not previously known.
13.3.1 Surfaces
The nature of adhesive interactions between clean, deformable metal tips indenting
metal surfaces have been identified and clarified over the course of the last decade
through the use of MD simulations [104,108–112]. In particular, the high surface
energies associated with clean metal surfaces can lead to strongly attractive inter-
actions between surfaces in contact. The strength of this attraction can be so large
that when the tip gets close enough to the surface to interact with it, surface atoms
“jump” upwards to wet the tip in a phenomenaknown as jump-to-contact(JC). This
wetting mechanism was first discovered in MD simulations [111] and has been con-
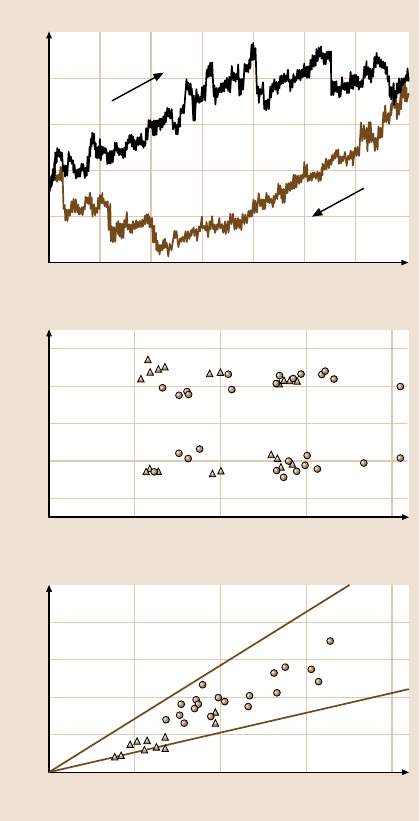
firmed experimentally [105,113–115] using the AFM, as shown in Fig. 13.2.
The MD simulations of Landman et al. [111,116–118] using EAM potentials
revealed that the JC phenomenon in metallic systems is driven by the need of the
atoms at the tip-surface interface to optimize their interaction energies while main-
taining their individual material cohesive binding. When the tip advances past the
JC point it indents the surface, which causes the force to increase. This behavior is
indicated in Fig. 13.3, points D to M. This region of the computer-generated force
curve has a maximum not present in the force curve generated from experimental
data (Fig. 13.3, point L). This is due to tip-induced flow of the metal atoms in the
surface that causes “pile-up” of the surface atoms around the edges of the indenter.
Hysteresis on the withdrawalof the tip, shown in Fig. 13.3, points M to X, is present
due to adhesion between the tip and the substrate. In particular, as the tip retracts
from the sample, a connective “neck” or nanowire of atoms forms between the tip
and the substrate that is primarily composed of metal atoms from the surface with
some atoms from the metal indenter that have diffused into the structure. A snapshot
from the MD simulations that illustrates this behavior is shown in Fig. 13.4.
As the tip is withdrawn farther, the magnitude of the force increases (becomes
more negative) until, at a critical force, the atoms in adjacent layers of the connec-
tive nanowire rearrange so that an additional row of atoms is created. This process
causes elongation of the connective nanowire and is responsible for the fine struc-
ture (apparent as a series of maxima) present in the retraction portion of the force
curve. These elongation and rearrangement steps are repeated until the connection
between the tip and the surface is broken. Similar elongation events have been ob-
served experimentally. For example, scanning tunneling microscopy (STM) exper-
666 Susan B. Sinnott et al.
0
150
Displacement (nm)
Force (nN)
0
0
Contact radius (nm)
k
eff
(N/m)
100
50
0,4 0.8 1.2
100
200
300
400
500
42
Pressure (GPa)
8
4
0
4
8
0
Contact radius (nm)
42
Fig. 13.2. Top: The experi-
mental values for the force
between a tip and a surface
that have a connective neck
between them. The neck con-
tracts and extends without
breaking on the scales shown.
Bottom:Theeffective spring
constant k
eff
determined ex-
perimentally for the connec-
tive necks and corresponding
maximum pressures, versus
contact radius of the tip. The
triangles indicate measure-
ments taken at room tem-
perature; the circles are the
measurements taken at liquid
He temperatures. After [115],
with permission of the ACS
(1996)
iments demonstrate that the metal nanowires between metal tips and surfaces can
elongate approximately 2500Å without breaking [119].
The JC process has been shown to affect the temperature at the tip–surface in-
terface. For instance, the constant-energy MD simulations of Tomagnini et al. [121]
predicted that the energy released due to the wetting of the tip by surface atoms
increases the temperature of the tip by about 15K at room temperature and is ac-
companied by significant structural rearrangement. At temperatures high enough to
cause the first few metal surface layers to be liquid, the distance at which the JC