Voet D., Voet Ju.G. Biochemistry
Подождите немного. Документ загружается.

Section 26-4. Amino Acids as Biosynthetic Precursors 1051
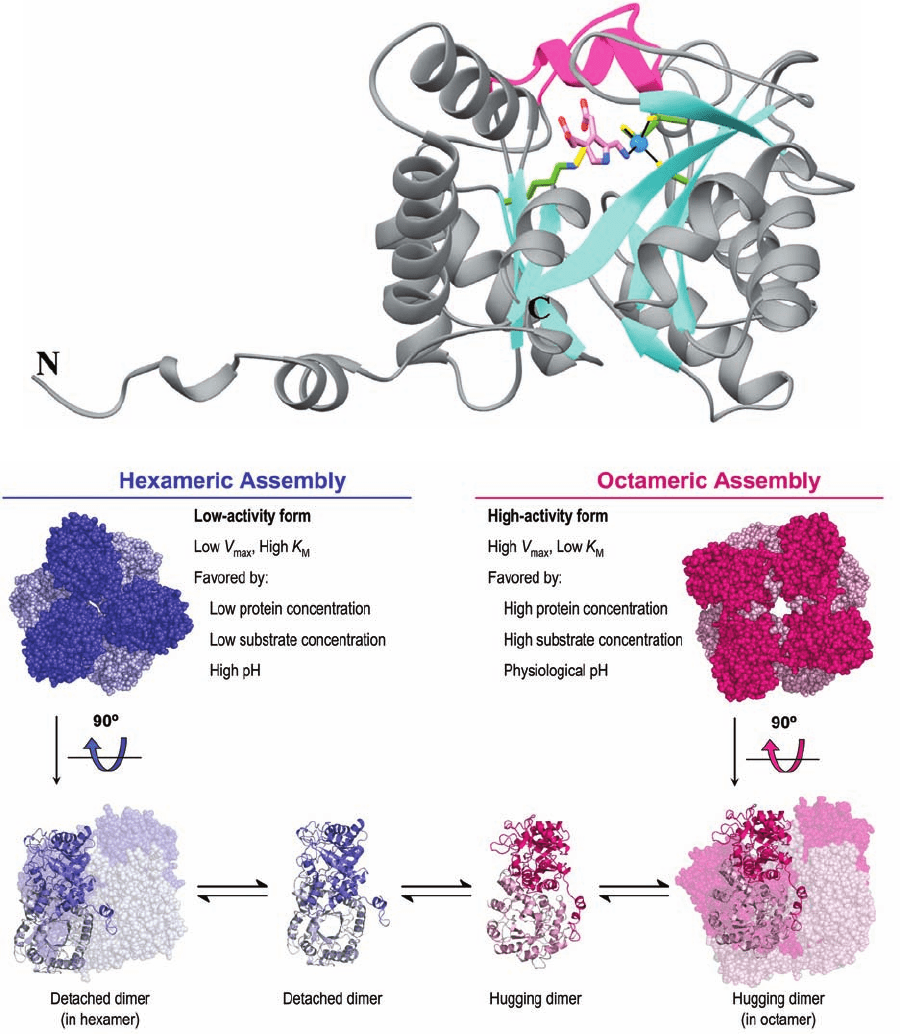
Figure 26-36 X-ray structure of human porphobilinogen
synthase (PBGS). (a) PBGS in covalent complex with its
porphobiligen (PBG) product.A monomer of this
homooctameric protein is viewed perpendicular to the axis of its
␣/ barrel and is drawn in gray with its  strands cyan and the
loop forming its flexible lid (residues 201–222) magenta.The
PBG, product, Lys 252 to which it is covalently linked, and the
three Cys side chains that ligand the active site Zn
2⫹
ion (blue
sphere) are shown in stick form with PBG C pink, side chain C
green, N blue, O red, S yellow, and the N¬C bond linking Lys
252 to PBG gold.The active site Zn
2⫹
ion is liganded (black
lines) by the S atoms of Cys 122, Cys 124, Cys 132, and the PBG
amino group. Lys 199, which lies directly behind Lys 252 in this
view, appears to be properly positioned to act as an acid–base
catalyst. [Based on an X-ray structure by Jonathan Cooper,
University of Southampton, U.K. PDBid 1E51.] (b) Quaternary
structural changes between the low activity hexameric state of
the F12L mutant of human PBGS (blue) and the high activity
octameric state of the wild-type enzyme (red). In the upper
panel, the proteins are viewed along their 3-fold and 4-fold axes
with the subunits closest to the viewer more darkly colored. In
the lower panel, the outer drawings are viewed along their 2-fold
axes with one dimer drawn in ribbon form and the others in
space-filling form.The inner drawings show only the dimers into
which the oligomers are assumed to dissociate before
reassembling to an alternate quaternary state. [Courtesy of Sarah
Lawrence and Eileen Jaffe,The Fox Chase Cancer Center,
Philadelphia, Pennsylvania. The X-ray structure of the F12L
mutant was determined by Eileen Jaffe. PDBid 1PV8.]
(a)
(b)
JWCL281_c26_1019-1087.qxd 4/20/10 9:26 AM Page 1051
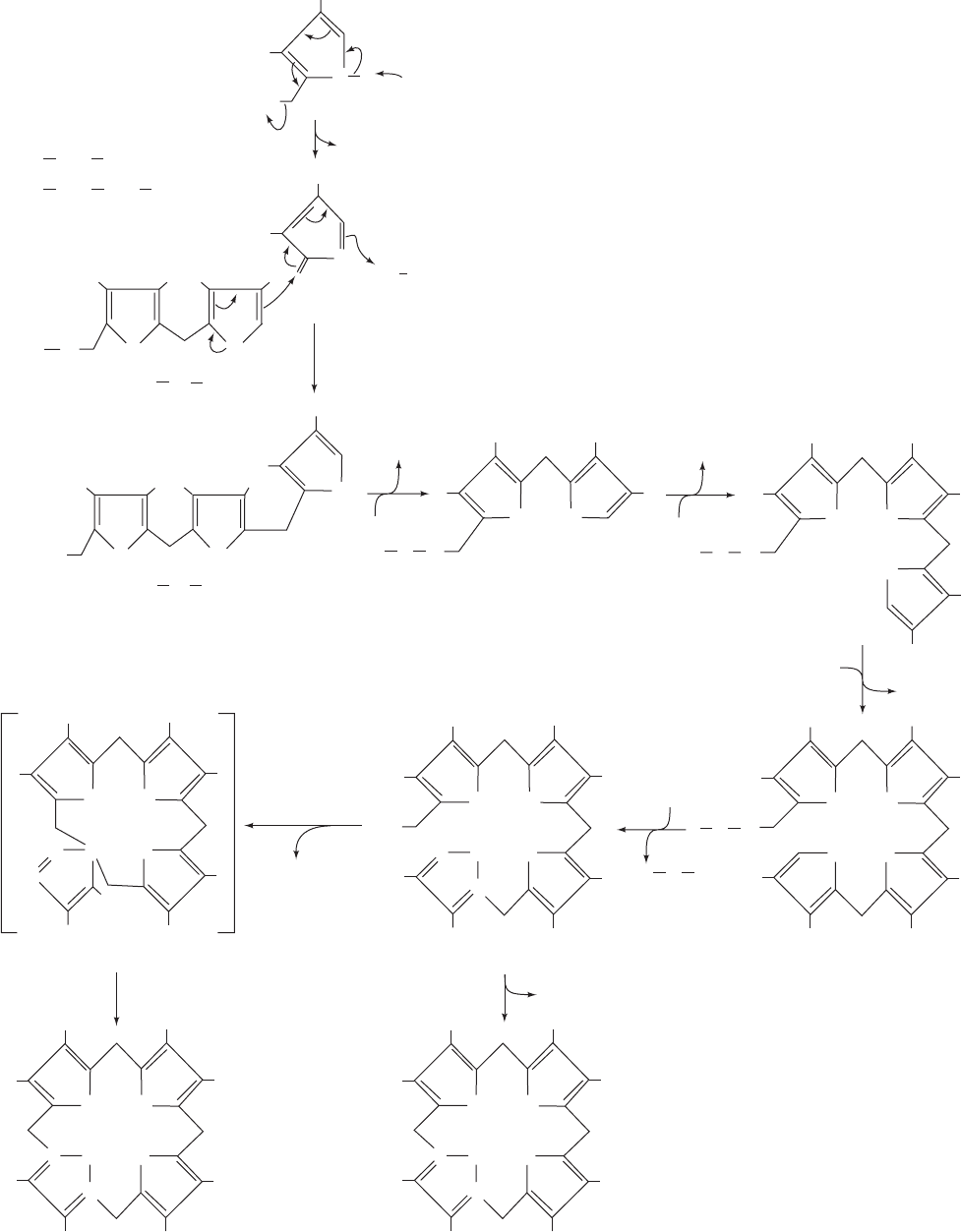
1052 Chapter 26. Amino Acid Metabolism
Figure 26-37 The synthesis of uroporphyrinogen III from
PBG as catalyzed by porphobilinogen deaminase and
uroporphyrinogen III synthase. (1a) General base-catalyzed
elimination of NH
3
to form a methylene pyrrolinene
intermediate. (1b) Addition to the methylene pyrrolinene
intermediate of the enzyme’s covalently linked dipyrromethane
cofactor to form a covalent adduct. (2–4) Sequential addition of
a second, third, and fourth PBG through successive NH
3
eliminations from PBG to form methylene pyrrolinene, as in
Reaction 1a, followed by addition of a pyrrole ring carbon atom
from the growing chain, as in Reaction 1b. (5) Hydrolysis of the
methylbilane–enzyme to yield hydroxymethylbilane and
regenerate the free enzyme–dipyrromethane complex.
(6) Synthesis of uroporphyrinogen III via a spiro intermediate by
porphobilinogen deaminase and uroporphyrinogen III synthase.
(7) Spontaneous cyclization of hydroxymethylbilane in the
absence of uroporphyrinogen III synthase. A and P represent
acetyl and propionyl groups.
A
P
N
H
A A
P
N
H
H
3
N
NH
3
C
1
C
1
A
P
N
H
C
2
C
2
B
+
1a
A
A
P
N
B
+
H
NH
3
A
A
A
P
NH
Methylene
pyrrolinene
S
Enz
E
C
1
C
2
E
C
1
C
2
E
C
1
C
2
E
A
P
N
H
C
1
AB
P
P
A
NH HN
A
AB
C
CD
BA
P
P
P
A
A
NH HN
NH
NH
HN
HN
HN
A
P
P
P
A
A
P
A
P
A
C
2
A
P
N
H
C
2
Enz
Enzyme–
dipyrromethane
1b
PBG
2
PBG
NH
3
NH
3
C
1
E
COO
–
CH
2
A =
COO
–
CH
2
P = CH
2
C
2
H
2
O
HO
C
1
E
3
PBG
H
2
O
5
4
PBG
6
C
D
BA
NH
NH
HN
HN
A
P
P
P
A
A
P
A
C
C
CD
BA
NH
NH
HN
HN
A
A
P
P
A
A
P
P
C
C
CD
BA
NH
NH
HN
HN
A
P
P
P
A
A
C
C
C
D
BA
NH HN
HN
A
A
P
P
A
A
P
P
C
N
C
H
2
O
7
uroporphy-
rinogen III
synthase
Hydroxymethylbilane
Methylbilane–enzyme
Spiro intermediate
Uroporphyrinogen III
Uroporphyrinogen I
6
uroporphyrinogen III
synthase
JWCL281_c26_1019-1087.qxd 4/20/10 9:26 AM Page 1052
d. The Porphyrin Ring Is Formed
from Four PBG Molecules
The next phase of heme biosynthesis is the condensation
of four PBG molecules to form uroporphyrinogen III, the
porphyrin nucleus, in a series of reactions catalyzed by por-
phobilinogen deaminase (alternatively, hydroxymethylbilane
synthase or uroporphyrinogen synthase) and uroporphyrino-
gen III synthase.The reaction (Fig. 26-37) begins with the en-
zyme’s displacement of the amino group in PBG to form a
covalent adduct. A second, third, and fourth PBG then se-
quentially add through the displacement of the primary
amino group on one PBG by a carbon atom on the pyrrole
ring of the succeeding PBG to yield a linear tetrapyrrole that
is hydrolyzed and released from the enzyme as hydrox-
ymethylbilane (also called preuroporphyrinogen).
e. Porphobilinogen Deaminase Has a
Dipyrromethane Cofactor
Peter Shoolingin-Jordan and Alan Battersby independ-
ently showed that porphobilinogen deaminase contains a
unique dipyrromethane cofactor (two pyrroles linked by a
methylene bridge; rings C
1
and C
2
in Fig. 26-37), which is co-
valently linked to the enzyme via a C¬S bond to an enzyme
Cys residue.Thus, the methylbilane–enzyme complex really
contains a linear hexapyrrole.The subsequent reaction step,
also catalyzed by porphobilinogen deaminase (Step 5 in Fig.
26-37), is the hydrolysis of the bond linking the second and
third pyrrole units of the hexapyrrole to yield hydroxy-
methylbilane and the dipyrromethane cofactor. This cofac-
tor is still linked to the enzyme, which is therefore ready to
catalyze a new round of hydroxymethylbilane synthesis.
How is the dipyrromethane cofactor assembled?
Shoolingin-Jordan has shown that porphobilinogen deami-
nase synthesizes its own cofactor from two PBG units us-
ing, it appears, the same catalytic machinery with which it
synthesizes methylbilane. However, the enzyme Cys reacts
much more rapidly with presynthesized hydroxymethylbi-
lane to form a reaction intermediate (the product of Step 2
in Fig. 26-37) that continues to add two more PBG units.
When hydroxymethylbilane is released,the enzyme retains
its dipyrromethane cofactor.
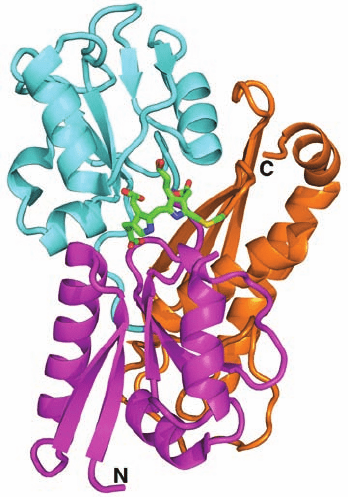
The X-ray structure of human porphobilinogen deaminase
(whose sequence is ⬎45% identical to those of the E. coli en-
zyme), in covalent complex with its dipyrromethane cofactor,
indicates that this monomeric, 364-residue protein folds into
three nearly equal sized domains (Fig 26-38). The
dipyrromethane cofactor lies deep in a cleft between domains
1 and 2 such that there is still considerable unoccupied space
in the cleft. Although the enzyme sequentially appends four
PBG residues to the cofactor, it has only one catalytic site.
If the enzyme has only one catalytic site, how does it
reposition the polypyrrole chain after each catalytic cycle
so that it can further extend this chain? One possibility is
that the polypyrrole chain fills the cavity next to the cofac-
tor. This model provides a simple steric rationale for why
the length of the polypyrrole chain is limited to six residues
(the final four of which are hydrolytically cleaved away by
the enzyme to yield the hydroxymethylbilane product and
regenerate the dipyrromethane cofactor).
f. Protoporphyrin IX Biosynthesis Requires
Four More Reactions
Cyclization of the hydroxymethylbilane product re-
quires uroporphyrinogen III synthase (Fig. 26-37). In the
absence of this enzyme, hydroxymethylbilane is released
from the synthase and rapidly cyclizes nonenzymatically to
the symmetric uroporphyrinogen I. Heme, however, is an
asymmetric molecule; the methyl substituent of pyrrole
ring D has an inverted placement compared to those of
rings A, B, and C (Fig. 26-32). This ring reversal to yield
uroporphyrinogen III has been shown by Battersby to pro-
ceed through attachment of the methylenes from rings A
and C to the same carbon of ring D so as to form a spiro
compound (a bicyclic compound with a carbon atom com-
mon to both rings; Fig. 26-37).
Heme biosynthesis takes place partly in the mitochon-
drion and partly in the cytosol (Fig. 26-39). ALA is mito-
chondrially synthesized and is transported to the cytosol for
conversion to PBG and then to uroporphyrinogen III. Pro-
toporphyrin IX, to which Fe is added to form heme, is pro-
duced from uroporphyrinogen III in a series of reactions
catalyzed by (1) uroporphyrinogen decarboxylase, which
decarboxylates all four acetate side chains (A) to form
methyl groups (M); (2) coproporphyrinogen oxidase, which
oxidatively decarboxylates two of the propionate side
chains (P) to vinyl groups (V); and (3) protoporphyrinogen
Section 26-4. Amino Acids as Biosynthetic Precursors 1053
Figure 26-38 X-ray structure of human porphobilinogen
deaminase in covalent complex with its dipyrromethane cofactor.
The protein is shown in ribbon form with domain 1 (residues
1–116 and 216–239) magenta, domain 2 (residues 117–215) cyan,
and domain 3 (residues 240–364) orange. The dipyrromethane
cofactor and the Cys side chain to which it is covalently linked are
drawn in stick form with C green, N blue, O red, and C yellow.
[Based on an X-ray structure by Zhi-Jie Liu, Institute of
Biophysics, Beijing, China. PDBid 3ECR.]
JWCL281_c26_1019-1087.qxd 8/9/10 9:40 AM Page 1053
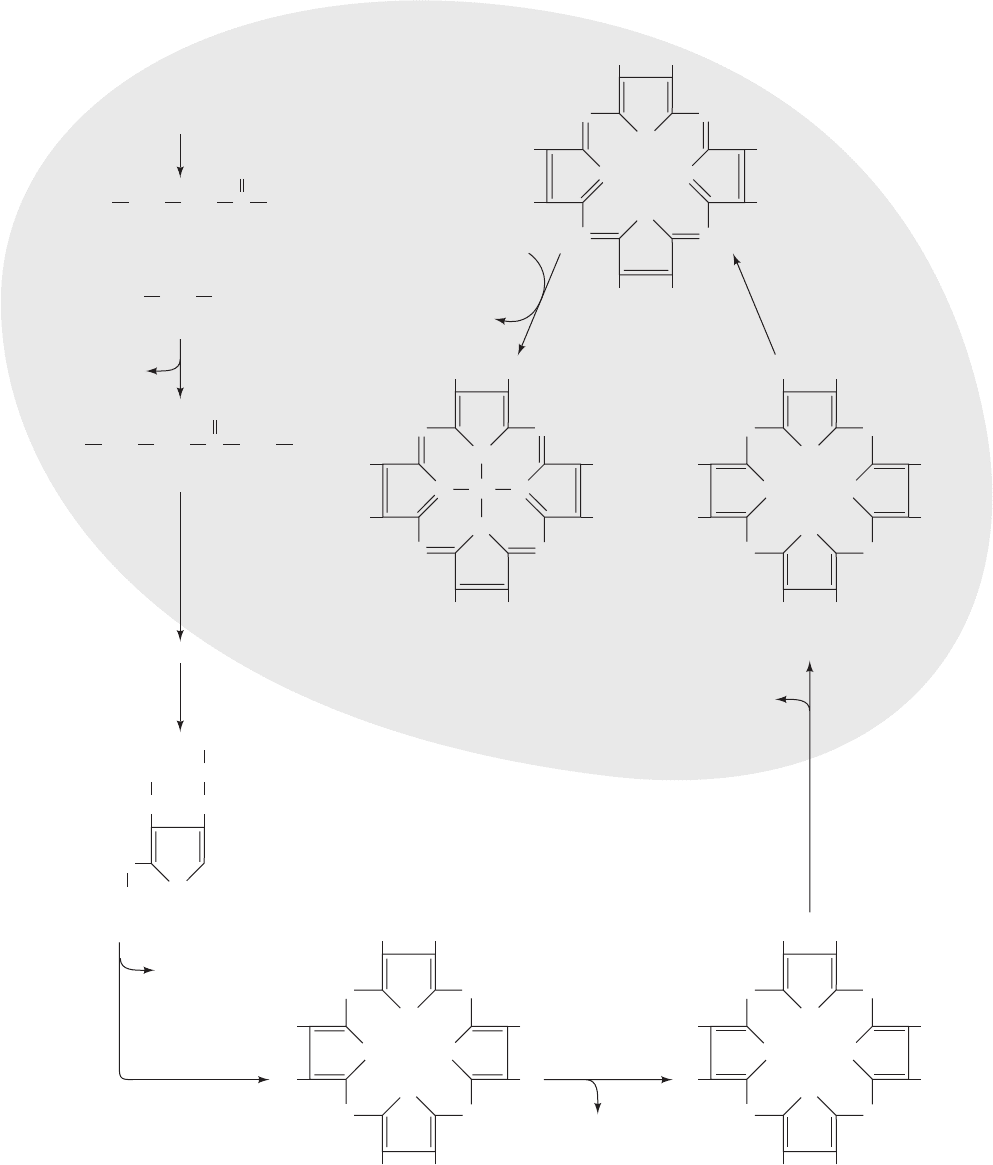
1054 Chapter 26. Amino Acid Metabolism
CH
2
Fe
N
N
NN
M V
P M
P
M
V
M
HC CH
HC CH
Heme
NH HN
M V
P M
P
M
V
M
H
2
C
Protoporphyrinogen IX
N
H
H
N
NN
M V
P M
P
M
V
M
HC CH
HC CH
Protoporphyrin IX
N
H
H
N
CH
2
H
2
C
C
O
CH
2
CH
2
CoA
–
OO
C
Succinyl-CoA
COO
–
H
3
N
+
CH
2
Glycine
+
C
O
CH
2
CH
2
–
OO
C
δ
-Aminolevulinic acid (ALA)
CH
2
NH
2
CO
2
δ-aminolevulinic
acid synthase
Fe
2+
2H
+
ferrochelatase
protoporphyrinogen
oxidase
2 CO
2
copropor-
phyrinogen
oxidase
CH
2
NH HN
M P
P M
P
M
P
M
H
2
C
Coproporphyrinogen III
N
H
H
N
CH
2
H
2
C
CH
2
NH HN
A P
P A
P
A
P
A
H
2
C
Uroporphyrinogen III
N
H
H
N
CH
2
H
2
C
uroporphyrinogen
decarboxylase
porphobilinogen
deaminase
uropor-
phyrinogen III
synthase
N
H
CH
2
CH
2
COO
–
H
2
C
–
OO
C
H
2
C
NH
2
ALA
porphobilinogen
synthase
Porphobilinogen (PBG)
4 CO
2
4 NH
3
Citric
acid
cycle
Mitochondrion
Cytosol
Figure 26-39 The overall pathway of heme biosynthesis.
␦-Aminolevulinic acid (ALA) is synthesized in the mitochondrion
by ALA synthase. ALA (left) leaves the mitochondrion and is
converted to PBG, four molecules of which condense to form a
porphyrin ring.The next two reactions involve oxidation of the
pyrrole ring substituents yielding protoporphyrinogen IX whose
formation is accompanied by its transport back into the
mitochondrion.After oxidation of the methylene groups linking
the pyrroles to yield protoporphyrin IX, ferrochelatase catalyzes
the insertion of Fe
2⫹
to yield heme. A, P, M, and V, respectively,
represent acetyl, propionyl, methyl,and vinyl (¬CH
2
“CH
2
) groups.
C atoms originating as the carboxyl group of acetate are red.
JWCL281_c26_1019-1087.qxd 6/8/10 9:38 AM Page 1054
oxidase, which oxidizes the methylene groups linking the
pyrrole rings to methenyl groups. Altogether, six carboxyl
groups originally from carboxyl-labeled acetate are lost as
CO
2
.The only remaining C atoms from carboxyl-labeled ac-
etate are the carboxyl groups of heme’s two propionate side
chains (P). During the coproporphyrinogen oxidase reac-
tion, the macrocycle is transported back into the mito-
chondrion for the pathway’s final reactions.
g. Ferrochelatase Catalyzes the Insertion of Fe(II)
Into Protoporphyrin IX to Form Heme
Protoporphyrin IX is converted to heme by the inser-
tion of Fe(II) into the tetrapyrrole nucleus by fer-
rochelatase, a protein that is associated with the inner mi-
tochondrial membrane on the matrix side. The X-ray
structure of human ferrochelatase in complex with its
heme product (Fig. 26-40), determined by Harry Dailey
and William Lanzilotta, reveals that the 369-residue sub-
units of this homodimeric protein consist of two struc-
turally similar domains and a C-terminal extension that oc-
curs only in animal ferrochelatases (Fig. 26-40). This
C-terminal extension participates in hydrogen bonding be-
tween the monomers; bacterial ferrochelatases, which lack
this extension, are monomeric although they are otherwise
structurally similar to the human enzyme despite their only
⬃10% sequence identity. In addition,the C-terminal exten-
sion is bound to the N-terminal domain by an unusual
[2Fe–2S] cluster that is coordinated by C196 of the
N-terminal domain and C403,C406, and C411 of the C-termi-
nal extension.The function of this [2Fe–2S] cluster, which is
distant from the active site, is unclear although it appears
likely that it only has a structural role. Nevertheless, three
mutations, C406Y, C406S, and C411G, that inactivate the
enzyme and thereby cause the rare inherited disease ery-
thropoietic protoporphyria (see below) demonstrate the
importance of the [2Fe–2S] cluster for activity.
The ferrochelatase active site (Fig. 26-40) consists of two
hydrophobic lips that, it has been proposed, participate in
the enzyme’s association with the inner mitochondrial
membrane. The ferrochelatase reaction follows an ordered
mechanism in which the Fe(II) binds to the enzyme before
the porphyrin. The reaction requires that the two pyrrole
NH protons be removed from the porphyrin prior to the
binding of the Fe(II) (Fig. 26-39). The invariant H263 ap-
pears properly positioned to abstract these protons from
the porphyrin (Fig. 26-40), a hypothesis that is supported
by mutagenesis studies. Structural and spectroscopic stud-
ies indicate that the metalation reaction is accompanied by
the folding of the porphyrin by ⬃12° to a nonplanar con-
formation. Product release is then facilitated by the partial
unwinding of a structurally conserved helix (Fig. 26-40;
helices are discussed in Section 8-1Bb).
h. Heme Biosynthesis Is Regulated Differently
in Erythroid and Liver Cells
The two major sites of heme biosynthesis are erythroid
cells, which synthesize ⬃85% of the body’s heme groups,
and the liver, which synthesizes ⬃80% of the remainder.
An important function of heme in liver is as the prosthetic
groups of the cytochromes P450, a family of oxidative en-
zymes involved in detoxification (Section 15-4Bc), whose
members are required throughout a liver cell’s lifetime in
amounts that vary with conditions. In contrast, erythroid
cells, in which heme is, of course,a hemoglobin component,
engage in heme synthesis only on differentiation, when
they synthesize hemoglobin in vast quantities.This is a one-
time synthesis; the heme must last the erythrocyte’s life-
time (normally 120 days) since heme and hemoglobin syn-
thesis stop on red cell maturation (protein synthesis stops
on the loss of nuclei and ribosomes). The different ways in
which heme biosynthesis is regulated in liver and in ery-
throid cells reflect these different demands: In liver, heme
biosynthesis must really be “controlled,” whereas in ery-
throid cells, the process is more like breaking a dam.
In liver, the main control target in heme biosynthesis is
ALA synthase, the enzyme catalyzing the pathway’s first
committed step. Heme, or its Fe(III) oxidation product
hemin, controls this enzyme’s activity through three mech-
anisms: (1) feedback inhibition, (2) inhibition of the trans-
port of ALA synthase (ALAS) from its site of synthesis in
the cytosol to its reaction site in the mitochondrion
(Fig. 26-39), and (3) repression of ALAS synthesis.
Section 26-4. Amino Acids as Biosynthetic Precursors 1055
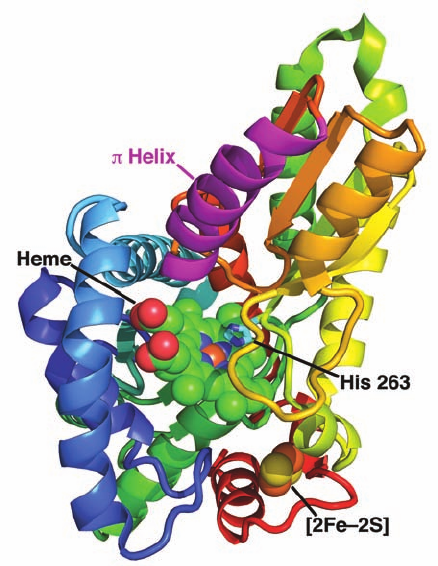
Figure 26-40 X-ray structure of human ferrochelatase in
complex with its heme product. A subunit of this homodimeric
protein is drawn in ribbon form colored in rainbow order from
its N-terminus (blue) to its C-terminus (red), but with its
conserved helix (residues 340–360) magenta.The bound heme
and [2Fe–2S] cluster are shown in space-filling form with C
green, N blue, O red, S yellow, and Fe orange.The side chain of
the catalytically essential His 263 is drawn in stick form with C
cyan and N blue. [Based on an X-ray structure by Harry Dailey
and William Lanzilotta, University of Georgia. PDBid 2QD3.]
JWCL281_c26_1019-1087.qxd 6/8/10 9:38 AM Page 1055
In erythroid cells, heme exerts quite a different effect on
its biosynthesis. Heme induces, rather than represses, pro-
tein synthesis in reticulocytes (immature erythrocytes).Al-
though the vast majority of the protein synthesized by
reticulocytes is globin, heme may also induce these cells to
synthesize the enzymes of the heme biosynthesis pathway.
Moreover, the rate-determining step of heme biosynthesis
in erythroid cells may not be the ALA synthase reaction,
which in mammalian reticulocytes is catalyzed by a differ-
ent isozyme (ALAS-2) than the ALA synthase that is ex-
pressed in other cells (ALAS-1). Experiments on various
systems of differentiating erythroid cells implicate fer-
rochelatase and porphobilinogen deaminase in the control
of heme biosynthesis in these cells. There are also indica-
tions that cellular uptake of iron may be rate limiting. Iron
is transported in the plasma complexed with the iron
transport protein transferrin. The rate at which the
iron–transferrin complex enters most cells, including those
of liver, is controlled by receptor-mediated endocytosis
(Section 12-5Bc). However, lipid-soluble iron complexes
that diffuse directly into reticulocytes stimulate in vitro
heme biosynthesis. The existence of several control points
supports the supposition that when erythroid heme biosyn-
thesis is “switched on,” all of its steps function at their maxi-
mal rates rather than any one step limiting the flow through
the pathway. Heme-stimulated synthesis of globin also en-
sures that heme and globin are synthesized in the correct ra-
tio for assembly into hemoglobin (Section 32-4Aa).
i. Porphyrias Have Bizarre Symptoms
Seven sets of genetic defects in heme biosynthesis, in
liver or erythroid cells, are recognized.All involve the accu-
mulation of porphyrin and/or its precursors and are there-
fore known as porphyrias (Greek: porphyra, purple). Two
such defects are known to affect erythroid cells: uropor-
phyrinogen III synthase deficiency (congenital erythropoi-
etic porphyria) and ferrochelatase deficiency (erythropoi-
etic protoporphyria). The former results in accumulation
of uroporphyrinogen I and its decarboxylation product
coproporphyrinogen I. Excretion of these compounds col-
ors the urine red, their deposition in the teeth turns them a
fluorescent reddish brown, and their accumulation in the
skin renders it extremely photosensitive such that it ulcer-
ates and forms disfiguring scars. Increased hair growth is
also observed in afflicted individuals such that fine hair
may cover much of the face and extremities. These symp-
toms have prompted speculation that the werewolf legend
has a biochemical basis.
The most common porphyria that primarily affects liver
is porphobilinogen deaminase deficiency (acute intermittent
porphyria). This disease is marked by intermittent attacks of
abdominal pain and neurological dysfunction, often brought
about by infection, fasting, certain drugs, alcohol, steroids,
and other chemicals, all of which induce the expression of
ALAS-1. Excessive amounts of ALA and PBG are excreted
in the urine during and after such attacks.The urine may be-
come red resulting from the excretion of excess porphyrins
synthesized from PBG in nonhepatic cells although the skin
does not become unusually photosensitive. King George III,
who ruled England during the American Revolution, and
who has been widely portrayed as being mad, in fact had at-
tacks characteristic of acute intermittent porphyria, was re-
ported to have urine the color of port wine, and had several
descendants who were diagnosed as having this disease.
American history might have been quite different had
George III not inherited this metabolic defect.
j. Heme Is Degraded to Bile Pigments
At the end of their lifetime, red cells are removed from the
circulation and their components degraded. Heme catabo-
lism (Fig.26-41) begins with oxidative cleavage, by heme oxy-
genase, of the porphyrin between rings A and B to form
biliverdin, a green linear tetrapyrrole. Biliverdin’s central
methenyl bridge (between rings C and D) is then reduced to
form the red-orange bilirubin. The changing colors of a heal-
ing bruise are a visible manifestation of heme degradation.
The highly lipophilic bilirubin is insoluble in aqueous
solutions. Like other lipophilic metabolites, such as free
fatty acids, it is transported in the blood in complex with
serum albumin. In the liver, its aqueous solubility is in-
creased by esterification of its two propionate side groups
with glucuronic acid, yielding bilirubin diglucuronide,
which is secreted into the bile. Bacterial enzymes in the
large intestine hydrolyze the glucuronic acid groups and, in
a multistep process, convert bilirubin to several products,
most notably urobilinogen. Some urobilinogen is reab-
sorbed and transported via the bloodstream to the kidney,
where it is converted to the yellow urobilin and excreted,
thus giving urine its characteristic color. Most of the uro-
bilinogen, however, is microbially converted to the deeply
red-brown stercobilin, the major pigment of feces.
When the blood contains excessive amounts of bilirubin,
the deposition of this highly insoluble substance colors the
skin and the whites of the eyes yellow.This condition, called
jaundice (French: jaune, yellow), signals either an abnor-
mally high rate of red cell destruction, liver dysfunction, or
bile duct obstruction. Newborn infants, particularly when
premature, often become jaundiced because their livers do
not yet make sufficient bilirubin UDP-glucuronosyltrans-
ferase to glucuronidate the incoming bilirubin.Jaundiced in-
fants are treated by bathing them with light from a fluores-
cent lamp; this photochemically converts bilirubin to more
soluble isomers that the infant can degrade and excrete.
k. Hemoglobin’s Reduced Affinity
for CO Prevents Asphyxiation
In the reaction forming biliverdin, the methenyl bridge
carbon between porphyrin rings A and B is released as CO
(Fig. 26-41, top), which, we have seen, is a tenacious heme
ligand (with 200-fold greater affinity for hemoglobin and
myoglobin than O
2
; Section 10-1A). Consequently, ⬃1% of
hemoglobin’s O
2
-binding sites are blocked by CO, even in
the absence of air pollution. However, free heme in solu-
tion binds CO with 20,000-fold greater affinity than it binds
O
2
. Thus, the globin (protein) portion of hemoglobin (and
likewise myoglobin) somehow lowers the affinity of its
bound heme for CO, thereby making O
2
transport possible.
How does the globin do so?
1056 Chapter 26. Amino Acid Metabolism
JWCL281_c26_1019-1087.qxd 4/20/10 9:26 AM Page 1056
Early X-ray structures of carboxymyoglobin (myoglo-
bin with a CO ligand) indicated that the bound CO was in-
clined from the normal to the heme plane by 40° to 60° (the
Fe¬C¬O bond angle appeared to be 120° to 140°), ap-
proximately the same angle with which O
2
binds to heme
(Fig. 10-12).Yet, in complexes of CO with porphyrins in the
absence of protein, the CO is normal to the heme plane.
This suggested that the globin (in both myoglobin and
hemoglobin) sterically bends the bound CO away from its
preferred linear geometry, thereby reducing its affinity for
CO and hence permitting the CO to be slowly exhaled.
However,a variety of spectroscopic investigations together
with highly accurate X-ray structures of carboxymyoglobin
revealed that the bound CO is, in fact, inclined from the
normal to the heme plane by ⬃7°, a distortion that is too
small to explain the reduced affinity of myoglobin for CO.
Section 26-4. Amino Acids as Biosynthetic Precursors 1057
Figure 26-41 The heme degradation pathway. M,V, P, and E, respectively, represent methyl,
vinyl, propionyl, and ethyl groups.
N
Fe
N
N
NN
M V
PM
P
M
V
M
Heme
A
C
DB
Fe
2+
O
2
+ CO +
N
M V
B
O
H
N
H
M P
C
M V
A
Biliverdin
Bilirubin
NADP
+
NADPH + H
+
Bilirubin diglucuronide
COO
–
OH
OH
HO
O
O
C O
CH
2
CH
2
OH
OH
HO
O
O
C O
CH
2
CH
2
bilirubin
UDP–glucuronosyl
transferase
(liver)
microbial
enzymes
(large intestine)
UDP UDP–glucuronate
H
2
O
Glucuronate
Urobilinogen
8H•
microbial enzymes
2H•
(kidney)
UrobilinStercobilin
2H•
microbial enzymes
(large intestine)
N
P M
D
N
H
N
M V
B
O
H
N
H
M P
C
M V
A
O
N
P M
D
N
M V
B
O
H H
N
H
M
C
M V
A
O
N
M
D
H H
H
C
H
N
H
H
H
H
H
H
H
N
H
N
M E
B
O
H
N
H
M P
C
M
A
O
N
P M
D
H H
H
HH
C
H
2
H H
N
H
N
M E
B
O
H
N
H
M P
C
M E
A
O
N
P M
D
HH
C
H
2
C
H
2
N
H
N
M E
B
O
H
N
H
M P
C
M E
A
O
N
P M
D
HH
C
H
2
C
H
2
HHH H
O
H
E
COO
–
JWCL281_c26_1019-1087.qxd 8/9/10 9:40 AM Page 1057
Of course, this reduced affinity might instead be explained
by the distortions that the upright CO ligand imposes on
the globin, presumably via the distal His (E7, the His
residue that hydrogen bonds to the bound O
2
; Section
10-2A). However, studies of the energetics of binding of
CO and O
2
to myoglobins in which His E7 has been mutated
to nonpolar residues of comparable bulk (e.g., Leu) indicate
that this is not the main determinant of the ligand affinity
changes. Rather, the reduction in affinity of myoglobin for
CO relative to that for O
2
has been shown to arise from the
greater hydrogen bonding affinity that His E7 has for O
2
rel-
ative to CO together with electrostatic effects due to the dif-
fering charge distributions in the O
2
and CO ligands.
l. Chloroquine Prevents Malaria by Inhibiting
Plasmodial Heme Sequestration
Malaria is caused by the mosquito-borne parasite Plas-
modium falciparum (Section 7-3Ab), which multiplies
within and destroys red blood cells in a 2-day cycle. During
the intraerythrocytic stages of its life cycle, the parasite par-
tially meets its nutritional needs by proteolyzing up to
⬃80% of the host cell’s hemoglobin in its so-called acid food
vacuole, whose pH is 4.7. This process releases heme, which
in its soluble form,is toxic to the parasite because it damages
cell membranes and inhibits a variety of enzymes. Since, un-
like their human hosts, plasmodia cannot degrade heme,
they sequester it within their food vacuoles in the form of
harmless dark brown granules known as hemozoin, which
consist of crystals of dimerized hemes linked together by re-
ciprocal iron–carboxylate bonds between the ferric ions
and the propionate side chains of adjacent molecules. He-
mozoin has been found to be identical to -hematin,
O
O
O
O
Fe(III)
Fe(III)
NN
N
N
NN
NN
O
HO
O
OH
-Hematin (hemozoin)
≥
whose X-ray structure has been determined. Dimers inter-
act in the crystals through hydrogen bonds between the re-
maining carboxyl groups.
Chloroquine,
a member of the quinoline ring–containing family of anti-
malarials, which includes quinine, is one of the most suc-
cessful antimicrobial agents that has been produced. It is
effective against plasmodia only during their intraerythro-
cytic stages. This drug, being a weak base that can readily
pass through membranes in its uncharged form, accumu-
lates in the plasmodial acid food vacuole in its acidic
(charged) form in millimolar concentrations. Chloroquine
and several other quinoline-containing antimalarials in-
hibit the crystallization of hemes to form hemozoin. This
inhibition in vivo is almost certainly responsible for the an-
timalarial properties of these drugs. The mechanism of in-
hibition is as yet unclear although a plausible hypothesis is
that the drug adsorbs onto crystallized hemozoin, inhibit-
ing further crystallization.
The massive use of chloroquine has, unfortunately, led
to the appearance of chloroquine-resistant plasmodia in
nearly every malarial region of the world. Resistant plas-
modia do not concentrate chloroquine in their food vac-
uoles to the high levels found in sensitive parasites.
Rather, they export this drug out of their food vacuoles at
an ⬃50-fold higher rate than do sensitive organisms. Since
chloroquine activity and chloroquine resistance have dif-
ferent mechanisms, it has been possible to modify existing
quinoline-containing structures and to develop new he-
mozoin crystallization inhibitors that are effective anti-
malarial agents but to which plasmodia are not (yet)
resistant.
B. Biosynthesis of Physiologically Active Amines
Epinephrine, norepinephrine, dopamine, serotonin (5-
hydroxytryptamine), ␥-aminobutyric acid (GABA), and
Cl
NH CH
CH
2
CH
2
CH
2
CH
3
CH
2
N(C
2
H
5
)
2
CH
N
H
3
CO
HO
N
N
Chloroquine
Quinine
1058 Chapter 26. Amino Acid Metabolism
JWCL281_c26_1019-1087.qxd 7/21/10 6:26 PM Page 1058
histamine
are hormones and/or neurotransmitters derived from amino
acids. For instance, epinephrine, as we have seen, activates
muscle adenylate cyclase, thereby stimulating glycogen
breakdown (Section 18-3E); deficiency in dopamine pro-
duction is associated with Parkinson’s disease, a degenera-
tive condition causing “shaking palsy”; serotonin causes
smooth muscle contraction; GABA is one of the brain’s
major inhibitory neurotransmitters (Section 20-5Cf), being
released at 30% of its synapses; and histamine is involved
in allergic responses (as allergy sufferers who take antihis-
tamines will realize), as well as in the control of acid secre-
tion by the stomach (Section 20-3C).
The biosynthesis of each of these physiologically active
amines involves decarboxylation of the corresponding pre-
cursor amino acid. Amino acid decarboxylases are PLP-
dependent enzymes that form a PLP–Schiff base with the
CCH
2
NH R
X
H
HO
HO
CH
2
CH
2
NH
3
+
HO
X
= OH, R = CH
3
Epinephrine (Adrenalin)
X
= OH, R = H Norepinephrine
X
= H, R = H Dopamine
Serotonin
(5-hydroxytryptamine)
–
OOC CH
2
CH
2
CH
2
NH
3
+
CH
2
CH
2
NH
3
+
γ-Aminobutyric acid (GABA)
N
NH
Histamine
N
substrate so as to stabilize the C
␣
carbanion formed on
C
␣
¬COO
⫺
bond cleavage (Section 26-1Aa):
Formation of histamine and GABA are one-step
processes (Fig. 26-42). In the synthesis of serotonin from
tryptophan, the decarboxylation is preceded by a hydroxy-
lation (Fig. 26-43) by tryptophan hydroxylase, one of three
mammalian enzymes that has a 5,6,7,8-tetrahydrobiopterin
cofactor (Section 26-3Ha). This hydroxylation involves an
NIH shift similar to that occurring in phenylalanine hy-
droxylase (Fig. 26-30), although no epoxide intermediate
has been observed in this case. Dopamine, norepinephrine,
and epinephrine are all termed catecholamines because
they are amine derivatives of catechol:
The conversion of tyrosine to these various catecholamines
occurs as follows (Fig. 26-44):
1. Tyrosine is hydroxylated to 3,4-dihydroxyphenylala-
nine (
L-DOPA) by tyrosine hydroxylase, another 5,6,7,8-
tetrahydrobiopterin-requiring enzyme.
2.
L-DOPA is decarboxylated to dopamine.
3. A second hydroxylation yields norepinephrine.
OH
OH
Catechol
H
H
R
C
C
O
C
...
H
O
–
O
–
N
2–
O
3
PO
α
N
H
CH
3
+
+
Section 26-4. Amino Acids as Biosynthetic Precursors 1059
Figure 26-42 The formation of ␥-aminobutyric acid (GABA) and histamine. The reactions
involve the decarboxylations of glutamate to form GABA and of histidine to form histamine.
CH
2
CH
2
Glutamate
NH
3
+
COO
–
CO
2
glutamate
decarboxylase
(PLP dependent)
C
H
–
OOC
CH
2
CH
2
CH
2
␥-Aminobutyric acid
(GABA)
NH
3
+
–
OOC
CH
2
Histidine
NH
3
+
COO
–
CO
2
histidine
decarboxylase
(PLP dependent)
C
H
CH
2
CH
2
Histamine
NH
3
+
N
H
N
H
N
N
H
N
H
N
JWCL281_c26_1019-1087.qxd 4/20/10 9:26 AM Page 1059
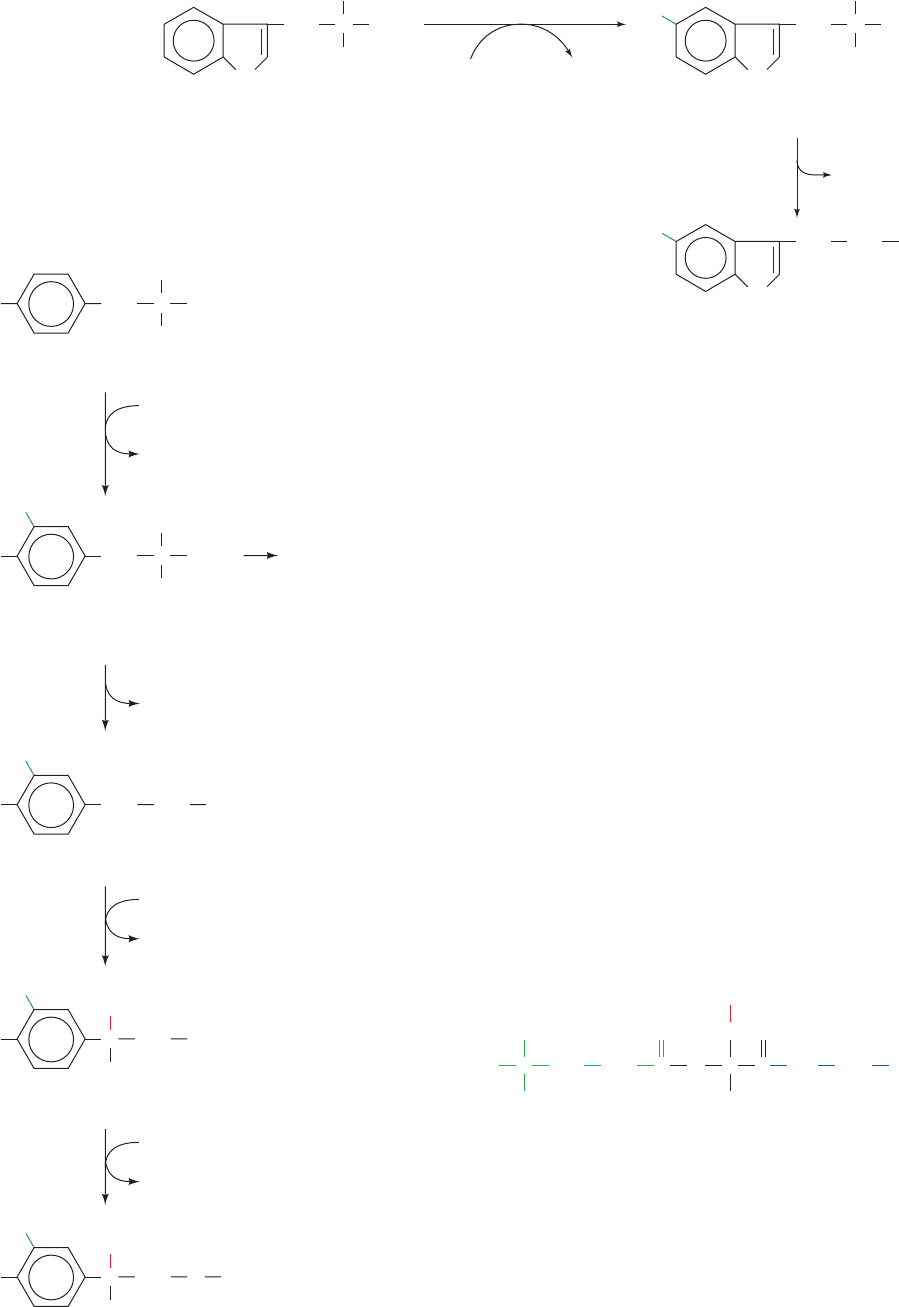
4. Methylation of norepinephrine’s amino group by
S-adenosylmethionine (SAM; Section 26-3Ea) produces
epinephrine.
The specific catecholamine that a cell produces depends
on which enzymes of the pathway are present. In adrenal
medulla, which functions to produce hormones (Section
19-1F), epinephrine is the predominant product. In some
areas of the brain, norepinephrine is more common. In
other areas, most prominently the substantia nigra, the
pathway stops at dopamine synthesis. Indeed, Parkinson’s
disease, which is caused by degeneration of the substantia
nigra, has been treated with some success by the admin-
istration of
L-DOPA, dopamine’s immediate precursor.
Dopamine itself is ineffective because it cannot cross the
blood–brain barrier.
L-DOPA, however, is able to get to its
sites of action where it is decarboxylated to dopamine. The
enzyme catalyzing this reaction, aromatic amino acid decar-
boxylase, decarboxylates all aromatic amino acids and is
therefore also responsible for serotonin formation.
L-DOPA
is also a precursor of the black skin pigment melanin.
C. Glutathione
Glutathione (GSH; ␥-glutamylcysteinylglycine),
a tripeptide that contains an unusual ␥-amide bond, partici-
pates in a variety of detoxification, transport, and metabolic
processes (Fig. 26-45). For instance, it is a substrate for
Glutathione
(GSH; ␥-glutamylcysteinylglycine)
N
H
O
C
O
C NHCH
2
CH
2
CH
2
H
2
C
H
3
N
C
H
COO
–
+
COO
–
SH
C
H
1060 Chapter 26. Amino Acid Metabolism
Tryptophan
CH
2
N
H
C
COO
–
NH
3
+
H
5-Hydroxytryptophan
CH
2
N
H
HO
C
COO
–
NH
3
+
H
Serotonin
CH
2
CO
2
aromatic amino
acid decarboxylase
(PLP dependent)
tryptophan
hydroxylase
Tetrahydro-
biopterin
+
O
2
Pterin-4a-
carbinolamine
N
H
HO
CH
2
NH
3
+
Figure 26-43 The
formation of serotonin.
The biosynthesis involves
the hydroxylation and
subsequent
decarboxylation of
tryptophan.
Figure 26-44 The sequential synthesis of
L-DOPA, dopamine, norepinephrine,
and epinephrine from tyrosine.
L-DOPA is also the precursor of the black skin
pigment melanin, an oxidized polymeric material.
CHO
Tyrosine
CH
2
COO
–
H
NH
3
+
Tetrahydrobiopterin + O
2
tyrosine
hydroxylase
Pterin-4a-carbinolamine
1
CHO
HO
Dihydroxyphenylalanine
(
L-DOPA)
CH
2
COO
–
H
OH
NH
3
+
Melanin
CO
2
aromatic amino
acid decarboxylase
2
HO
HO
Dopamine
CH
2
CH
2
NH
3
+
O
2
+ Ascorbate
dopamine
-hydroxylase
H
2
O + Dehydroascorbate
3
HO
HO
Norepinephrine
C
H
CH
2
NH
3
+
S-Adenosylmethionine
phenylethanolamine
N
-methyltransferase
S-Adenosylhomocysteine
4
OH
HO
HO
Epinephrine
C
H
CH
2
CH
3
N
H
JWCL281_c26_1019-1087.qxd 4/20/10 9:26 AM Page 1060