Voet D., Voet Ju.G. Biochemistry
Подождите немного. Документ загружается.

H
C
CH
2
N
H
COO
–
NH
3
+
Tryptophan
H
C
CH
2
N
H
COO
–
NH
3
+
-Formylkynurenine
C
O
CH
O
N
H
C
CH
2
COO
–
NH
3
+
Kynurenine
C
O
NH
2
2-Amino-3-carboxymuconate-
6-semialdehyde
H
C
H
3
C COO
–
NH
3
+
3-Hydroxyanthranilate
H
C
CH
2
COO
–
NH
3
+
3-Hydroxykynurenine
C
O
NH
2
OH
COO
–
NH
2
OH
Alanine
+
COO
–
NH
2
–
OOC
OCH
Quinolinate
COO
–
N
COO
–
2-Aminomuconate-
6-semialdehyde
H
NH
2
–
OOC
OCH
2-Aminomuconate
NH
2
–
OOC
–
OOC
α
-Ketoadipate
O
–
OOC
–
OOC
CH
2
COO
–
C
O
CH
3
Acetoacetate
O
2
HCOO
–
H
2
O
H
2
O + NADP
+
O
2
+ NADPH
H
2
O
O
2
H
2
O
spontaneous
To NAD
+
and NADP
+
NADH
NAD
+
CO
2
NAD(P)
+
NAD(P)H
H
2
O
101112131415
98
7
6
5
4
3
21
NH
3
+
16
CO
2
CO
2
and ␣-ketoglutarate dehydrogenase (Sections 21-2A and
21-3D). Reactions 6, 8, and 9 are standard reactions of fatty
acyl-CoA oxidation: dehydrogenation by FAD, hydration,
and dehydrogenation by NAD
⫹
. Reactions 10 and 11 are
standard reactions in ketone body formation. Two mole-
cules of CO
2
are produced at Reactions 5 and 7 of the
pathway.
The saccharopine pathway is thought to predominate in
mammals because a genetic defect in the enzyme that cat-
alyzes Reaction 1 in the sequence results in hyperlysinemia
and hyperlysinuria (elevated levels of lysine in the blood and
urine, respectively) along with mental and physical retarda-
tion.This is yet another example of how the study of rare in-
herited disorders has helped to trace metabolic pathways.
Leucine’s carbon skeleton,as we have seen, is converted
to one molecule each of acetoacetate and acetyl-CoA,
whereas that of lysine is converted to one molecule of ace-
toacetate and two of CO
2
. Since neither acetoacetate nor
acetyl-CoA can be converted to glucose in animals, leucine
and lysine are purely ketogenic amino acids.
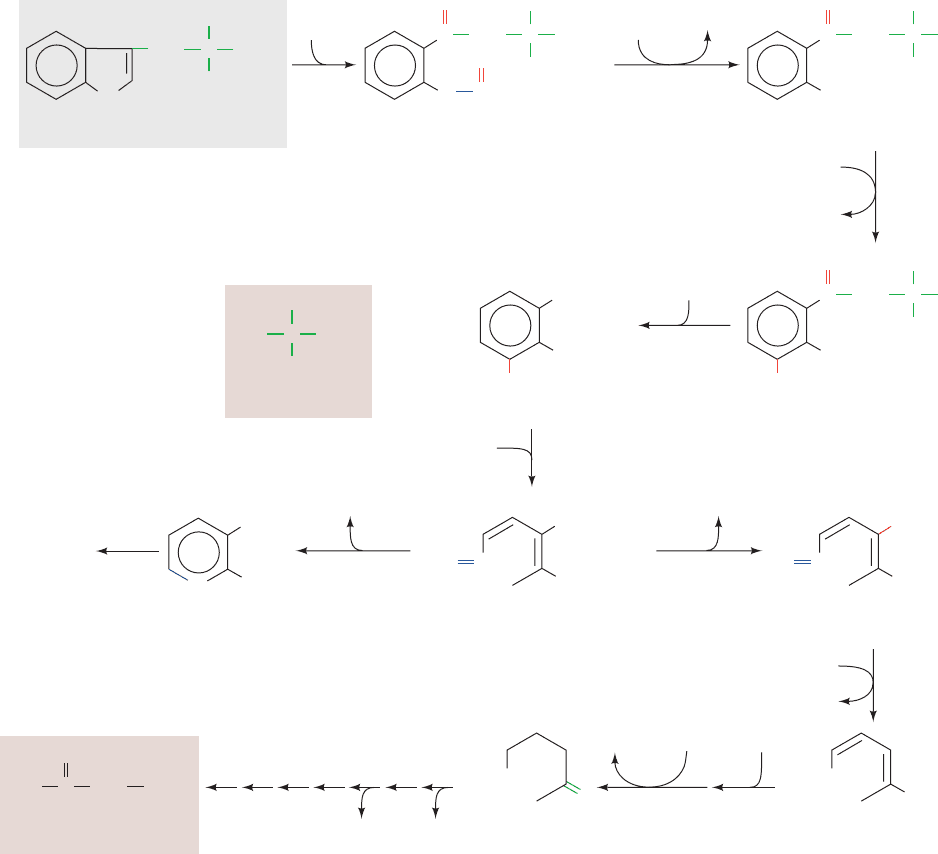
G. Tryptophan Is Degraded to Alanine
and Acetoacetate
The complexity of the major tryptophan degradation path-
way (Fig. 26-24) precludes detailed discussion of all of its
reactions. However,one reaction in the pathway is of partic-
ular interest. Reaction 4, cleavage of 3-hydroxykynurenine
Section 26-3. Metabolic Breakdown of Individual Amino Acids 1041
Figure 26-24 The pathway of tryptophan degradation. The
enzymes involved are (1) tryptophan-2,3-dioxygenase,
(2) formamidase, (3) kynurenine-3-monooxygenase,
(4) kynureninase (PLP dependent), (5) 3-hydroxyanthranilate-
3,4-dioxygenase, (6) amino carboxymuconate semialdehyde
decarboxylase, (7) aminomuconate semialdehyde dehydrogenase,
(8) hydratase, (9) dehydrogenase, and (10–16) enzymes of
Reactions 5 through 11 in lysine degradation (Fig. 26-23).
2-Amino-3-carboxymuconate-6-semialdehyde, in addition to
undergoing Reaction 6, spontaneously forms quinolinate, an
NAD
⫹
and NADP
⫹
precursor (Section 28-5A).
JWCL281_c26_1019-1087.qxd 4/20/10 9:26 AM Page 1041
H
C
CH
2
COO
–
NH
3
+
3-Hydroxykynurenine
C
O
NH
2
OH
H
C
CH
2
COO
–
+
NH
H
+
C
O
NH
2
1
2
3
4
6
5
CH
E
X
+
N
H
+
+
NH
P
+
N
H
OH
+
P
p
E
X
NH
3
+
p
+
Enzyme–PLP Schiff base
•
C
CH
2
COO
–
+
NH
C
O
NH
2
CH
3
N
H
OH
OH
P
E
X
+
NH
3
p
p
•
C
CH
2
COO
–
+
NH
C
O
–
NH
2
N
H
OH
P
E
X
+
+
NH
3
C
H
2
C
COO
–
O
–
)NH
C
O
H
2
O
N
H
H
+
OH
P
E
X
+
+
NH
3
E
X
+
NH
3
p
E
X
+
NH
3
p
E
X
NH
+
+
C
H
H
3
C
COO
–
+
NH
N
H
P
N
H
P
C
H
3
C
COO
–
+
NH
H
+
N
H
P
Acyl–enzyme
C
O
OH
3-Hydroxy-
anthranilate
7
8
•
C
3
H
H
3
C
COO
–
NH
+
+
Alanine
Enzyme–PLP
Schiff base
+
+
OH
CH
3
OH
CH
3
OH
CH
3
OH
CH
3
OH
CH
3
NH
2
NH
2
OH
CH
3
OH
CH
3
␥

␣
CH
CH
CH
CH
CH
CH
CH
..
p
..
..
..
..
..
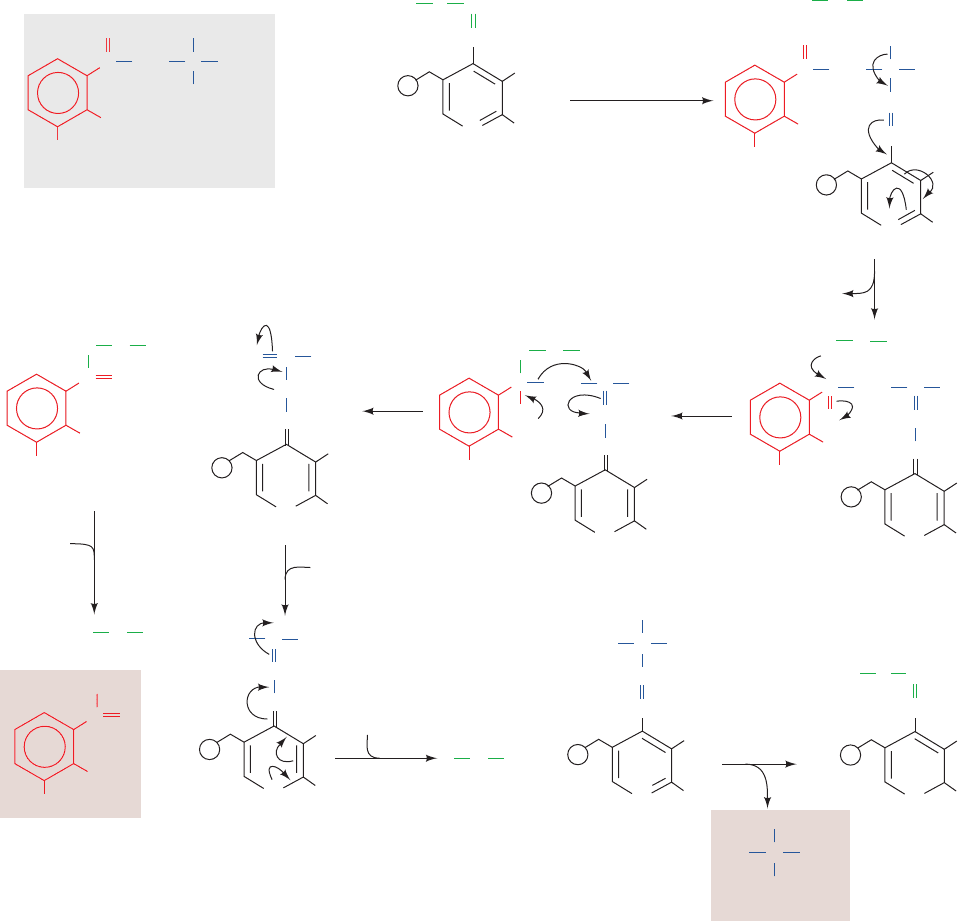
to alanine and 3-hydroxyanthranilate, is catalyzed by
kynureninase, a PLP-dependent enzyme. The reaction fur-
ther demonstrates the enormous versatility of PLP. We
have seen how PLP can labilize an ␣-amino acid’s C
␣
¬H
and C
␣
¬C

bonds (Fig. 26-16). Here we see the facilitation
of C

¬C
␥
bond cleavage. The reaction follows the same
steps as transamination reactions but does not hydrolyze
the tautomerized Schiff base (Fig. 26-25).The proposed re-
action mechanism involves an attack of an enzyme nucle-
ophile on the carbonyl carbon (C
␥
) of the tautomerized
3-hydroxykynurenine–PLP Schiff base (Fig. 26-25, Step 3).
This is followed by C

¬C
␥
bond cleavage to generate an
acyl–enzyme intermediate together with a tautomerized
alanine–PLP adduct (Fig. 26-25, Step 4). Hydrolysis of the
acyl–enzyme then yields 3-hydroxyanthranilate, whose fur-
ther degradation yields ␣-ketoadipate (Fig. 26-24, Reac-
tions 5–9). ␣-Ketoadipate is also an intermediate in lysine
breakdown (Fig. 26-23, Reaction 4) so that the last seven
reactions in the degradation of both these amino acids are
identical, forming acetoacetate and two molecules of CO
2
.
1042 Chapter 26. Amino Acid Metabolism
Figure 26-25 Proposed mechanism for the PLP-dependent
kynureninase-catalyzed C

¬C
␥
bond cleavage of
3-hydroxykynurenine. The reaction occurs in eight steps:
(1) transimination; (2) tautomerization; (3) attack of an enzyme
nucleophile, X; (4) C

¬C
␥
bond cleavage with formation of an
acyl–enzyme intermediate; (5) acyl–enzyme hydrolysis; (6) and
(7) tautomerization; and (8) transimination.
JWCL281_c26_1019-1087.qxd 6/8/10 9:38 AM Page 1042
CHHO
HO
OH
O
Tyrosine
CH
2
COO
–
NH
3
+
C
p-Hydroxyphenylpyruvate
CH
2
COO
–
O
2
CO
2
α-Ketoglutarate
Glutamate
2
3
Tetrahydrobiopterin + O
2
Dihydrobiopterin + H
2
O
1
C
CH
COO
–
COO
–
H
O
C
O
C
HO
Homogentisate
CH
2
CH
2
CH
2
COO
–
O
2
H
2
O
4
5
4-Maleylacetoacetate
C
CH
–
OOC
COO
–
H
O
C
O
C
CH
2
CH
2
6
4-Fumarylacetoacetate
+
Fumarate
COO
–
C
CH
–
OOC
H
Acetoacetate
O
CCH
2
CH
3
CH
Phenylalanine
CH
2
COO
–
NH
3
+
COO–
x
*
⌬
*
*
*
*
*
⌬
⌬
⌬
⌬
⌬
*
⌬x
x
x
x
x
x
NN
N
N
Pteridine
N
H
O
O
NN
N
H
C B A
Isoalloxazine
N
H
O
NN
R⬘
CH
3
CH
3
O
N
Flavin
O
N
N
H
NN
Pterin
(2-amino-4-oxopteridine)
R
1
2
3
4
5
6
7
8
Biopterin: R =
H
C
C
HO
H
OH
CH
3
O
C
–
OOC
N
H
N
H
C
H
COO
–
C
H
H
C
H
H
C
H
H
Folate: R =
H
2
N
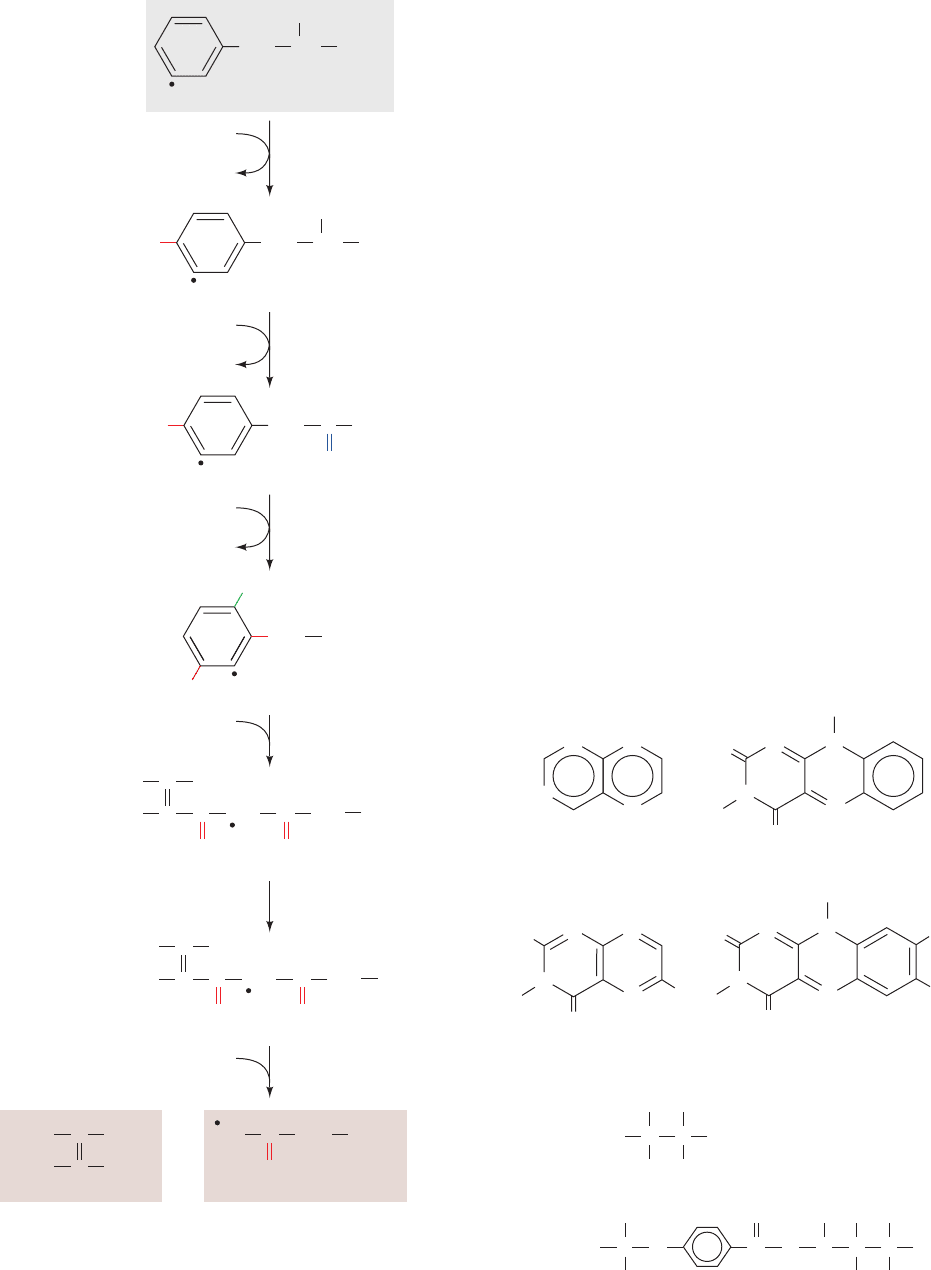
H. Phenylalanine and Tyrosine Are Degraded
to Fumarate and Acetoacetate
Since the first reaction in phenylalanine degradation is its
hydroxylation to tyrosine, a single pathway (Fig. 26-26) is
responsible for the breakdown of both of these amino
acids.The final products of the six-reaction degradation are
fumarate, a citric acid cycle intermediate, and acetoacetate,
a ketone body.
a. Pterins Are Redox Cofactors
The hydroxylation of phenylalanine by the nonheme
iron–containing homotetrameric enzyme phenylalanine
hydroxylase (PAH) requires O
2
and that the iron be in the
Fe(II) state. The enzyme also requires the participation of
biopterin, a pterin derivative. Pterins are compounds that
contain the pteridine ring (Fig. 26-27). Note the resem-
blance between the pteridine ring and the isoalloxazine
ring of the flavin coenzymes; the positions of the nitrogen
atoms in pteridine are identical with those of the B and C
rings of isoalloxazine. Folate derivatives also contain the
pterin ring (Section 26-4D). Pterins, like flavins, participate
in biological oxidations. The active form of biopterin is the
fully reduced form, 5,6,7,8-tetrahydrobiopterin (BH
4
). It is
produced from 7,8-dihydrobiopterin and NADPH, in what
Section 26-3. Metabolic Breakdown of Individual Amino Acids 1043
Figure 26-26 The pathway of phenylalanine degradation. The
enzymes involved are (1) phenylalanine hydroxylase,
(2) aminotransferase, (3) p-hydroxyphenylpyruvate dioxygenase,
(4) homogentisate dioxygenase, (5) maleylacetoacetate
isomerase, and (6) fumarylacetoacetase. The symbols labeling the
various carbon atoms serve to indicate the group migration that
occurs in Reaction 3 of the pathway (see Fig. 26-31).
Figure 26-27 The pteridine ring, the nucleus of biopterin
and folate. Note the similar structures of pteridine and the
isoalloxazine ring of flavin coenzymes.
JWCL281_c26_1019-1087.qxd 4/20/10 9:26 AM Page 1043
3
H
O
N
N
H
NN
5
6
7
8
H
C
C
HO
OH
H
CH
3
H
H
H
H
2
N
7,8-Dihydrobiopterin
O
N
N
H
NN
5
6
7
8
H
C
C
HO
H
OH
CH
3
H
H
H
H
2
N
5,6,7,8-Tetrahydrobiopterin (BH
4
)
H
H
O
N
N
NN
H
C
C
HO
H
H
OH
CH
3
H
H
H
H
2
N
H
2
O
HO
H
O
N
N
H
NN
H
C
C
HO
H
OH
CH
3
H
H
H
HN
7,8-Dihydrobiopterin
(quinoid form)
3
H
H
C
CH
2
COO
–
NH
3
+
HH
HH
+
O
2
Phenylalanine
H
C
CH
2
COO
–
NH
3
+
HH
H
Tyrosine
HO
phenylalanine
hydroxylase
pterin-4a-
carbinolamine
dehydratase
dihydropteridine
reductase
NAD(P)H
NAD(P)
+
NADP
+
NADPH + H
+
dihydrofolate reductase
H
Pterin-4a-carbinolamine
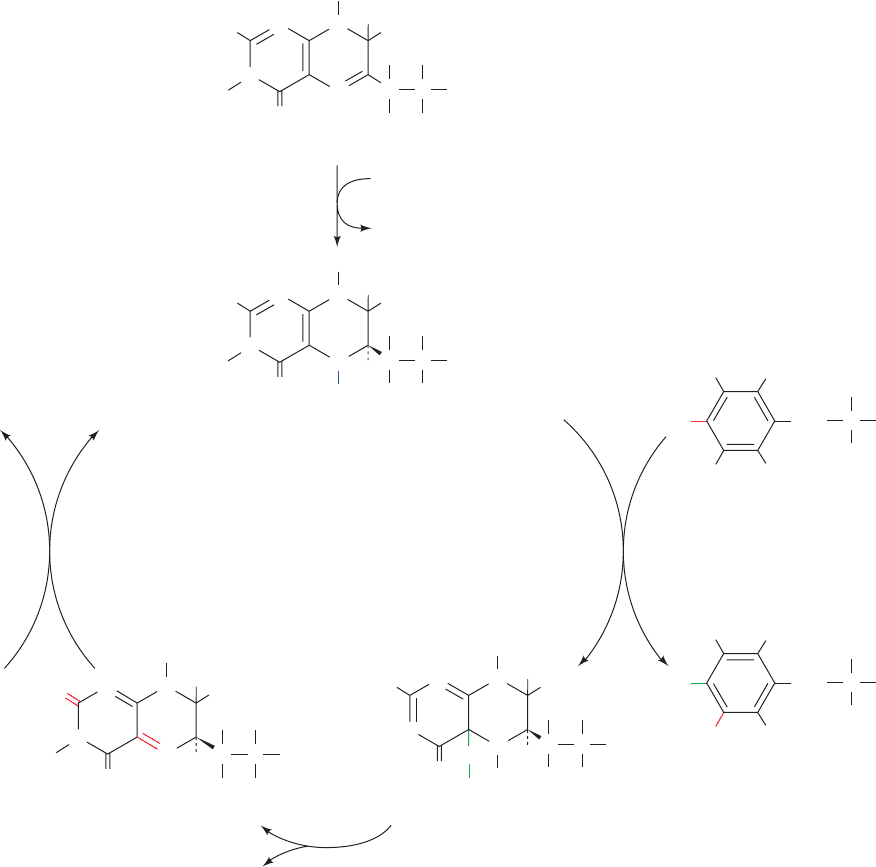
may be considered a priming reaction, by dihydrofolate
reductase (Fig. 26-28).
Each 452-residue subunit of the PAH homotetramer
contains three domains, an N-terminal regulatory do-
main, a catalytic domain, and a C-terminal tetrameriza-
tion domain. However, the 325-residue catalytic domain
alone forms catalytically competent dimers. The X-ray
structure of the catalytic domain of PAH in its Fe(II) state
in complex with BH
4
, determined by Edward Hough, re-
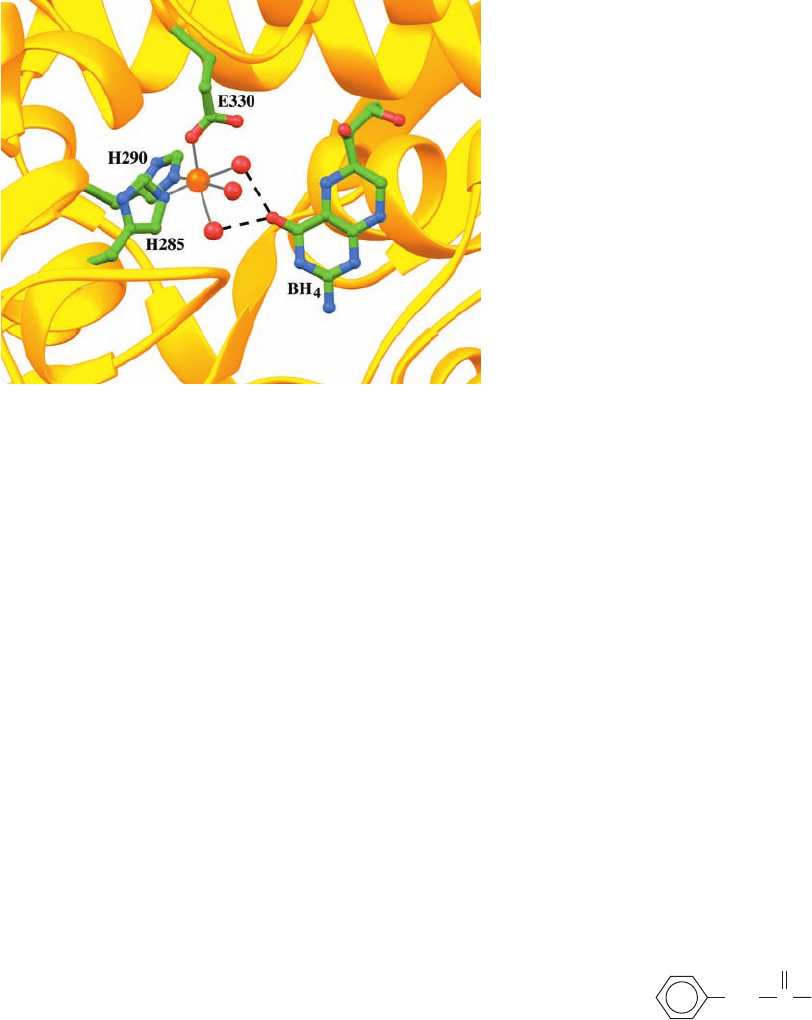
veals that the Fe(II) is octahedrally coordinated by His
285, His 290, Glu 330, and three water molecules, and that
atom O4 of BH
4
is hydrogen bonded to two of these
waters (Fig. 26-29).
In the phenylalanine hydroxylase reaction, 5,6,7,8-
tetrahydrobiopterin is hydroxylated to pterin-4a-carbinol-
amine (Fig. 26-28), which is converted to 7,8-dihydro-
biopterin (quinoid form) by pterin-4a-carbinolamine
dehydratase. The quinoid is subsequently reduced by the
NAD(P)H-requiring enzyme dihydropteridine reductase
to regenerate the active cofactor. Note that although
dihydrofolate reductase and dihydropteridine reductase
produce the same product, they utilize different tau-
tomers of the substrate.Although this suggests that these
enzymes may be evolutionarily related, the comparison
of their X-ray structures indicates that this is not the
case: Dihydropteridine reductase resembles nicotinamide
1044 Chapter 26. Amino Acid Metabolism
Figure 26-28 The formation, utilization, and regeneration of 5,6,7,8-tetrahydrobiopterin (BH
4
)
in the phenylalanine hydroxylase reaction.
JWCL281_c26_1019-1087.qxd 6/8/10 9:38 AM Page 1044
coenzyme–requiring flavin-dependent enzymes such as
glutathione reductase and dihydrolipoyl dehydrogenase
(Section 21-2B).
b. Phenylalanine Hydroxylase Is Controlled by
Phosphorylation and by Allosteric Interactions
PAH initiates the detoxification of high concentrations
of phenylalanine as well as the synthesis of the cate-
cholamine hormones and neurotransmitters (Section 26-4B).
It is allosterically activated by its substrate, phenylalanine,
and by phosphorylation at its Ser 16 by the cAMP-dependent
protein kinase A (PKA; Section 18-3Cb). Its second sub-
strate, BH
4
, allosterically inhibits the enzyme.
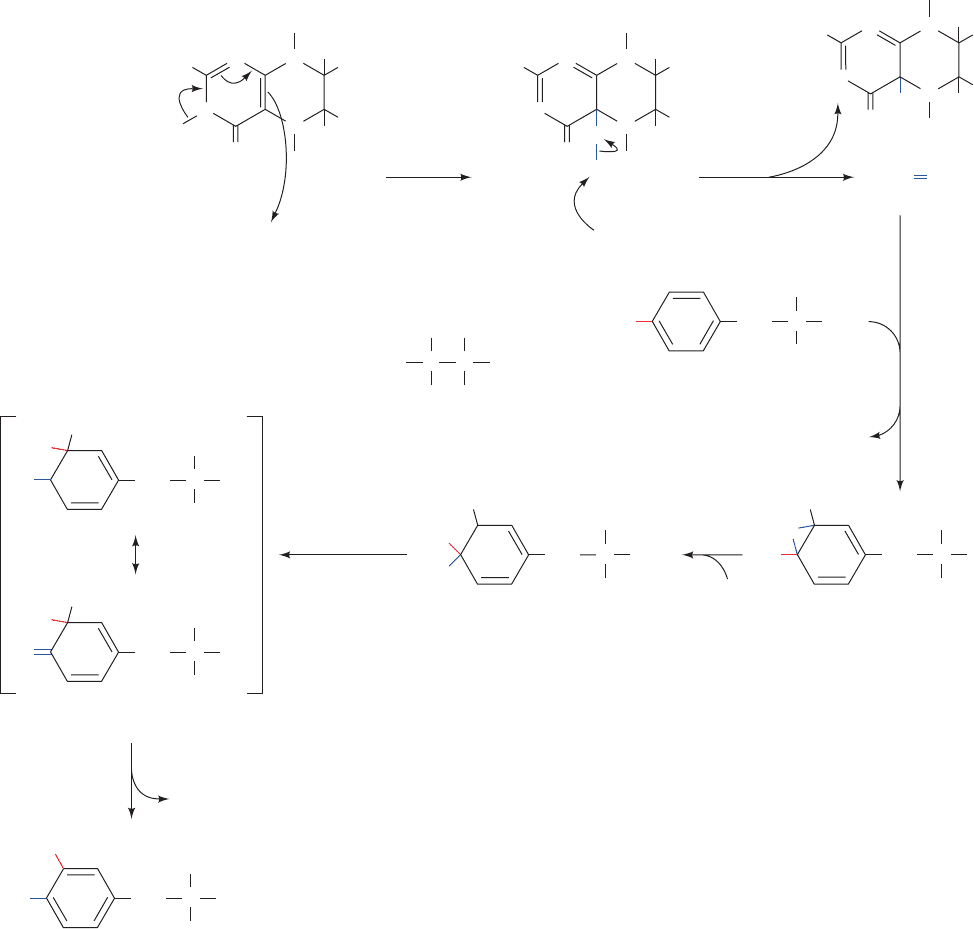
c. The NIH Shift
An unexpected aspect of the PAH reaction is that a
3
H
atom, which begins on C4 of phenylalanine’s phenyl ring,
ends up on C3 of this ring in tyrosine (Fig. 26-28, right)
rather than being lost to the solvent by replacement with
the OH group. The mechanism postulated to account for
this NIH shift (so called because it was first characterized
by chemists at the National Institutes of Health) involves
the activation of oxygen by the pterin and Fe cofactors to
form the pterin-4a-carbinolamine and a reactive oxyferryl
group [Fe(IV)“O
2⫺
; Fig. 26-30, Steps 1 and 2] that reacts
with the substrate to form an epoxide across the phenyl
ring’s 3,4 bond (Fig. 26-30, Step 3).This is followed by epox-
ide opening to form a carbocation at C3 (Fig.26-30, Step 4).
Migration of a hydride from C4 to C3 forms a more stable
carbocation (an oxonium ion; Fig. 26-30, Step 5). This mi-
gration is followed by ring aromatization to form tyrosine
(Fig. 26-30, Step 6). Tyrosine hydroxylase and tryptophan
hydroxylase (Section 26-4B) are both homologous to
phenylalanine hydroxylase and utilize this same NIH shift
reaction mechanism, although there may not be an epoxide
intermediate in these cases.
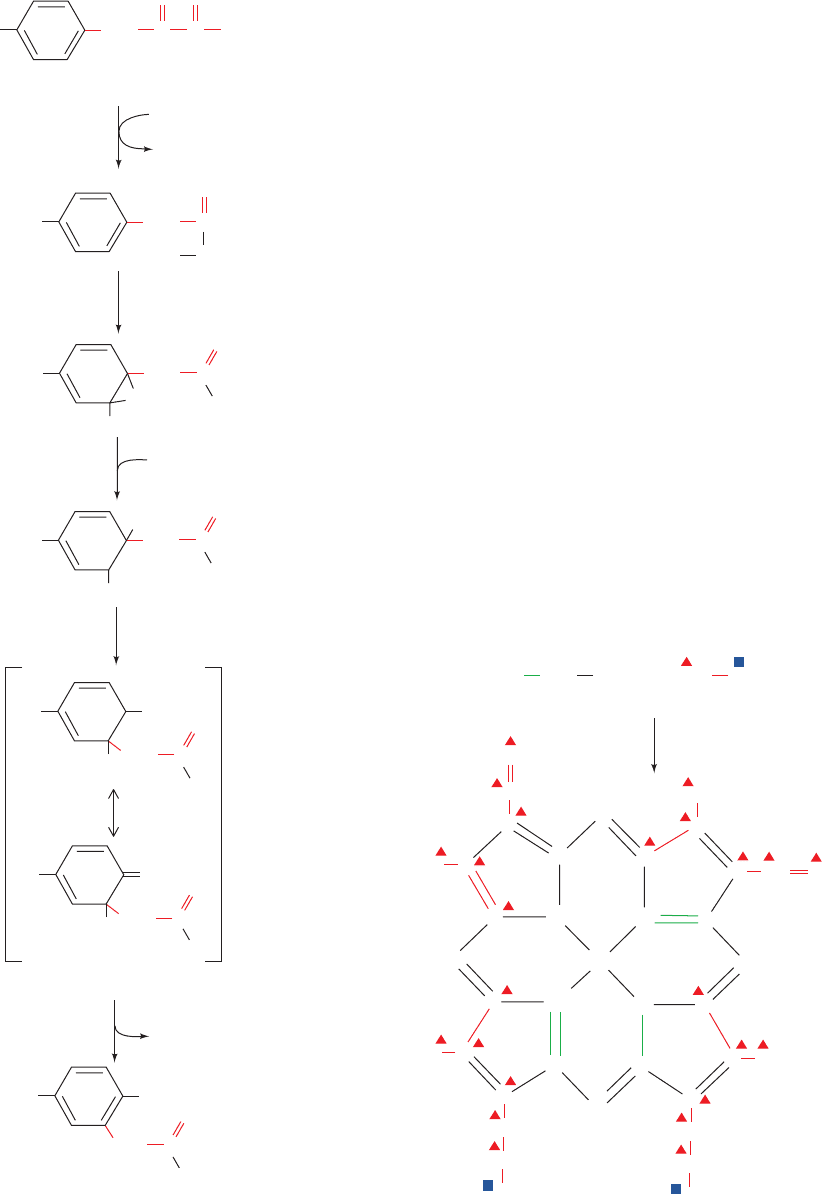
Reaction 3 in the phenylalanine degradation pathway
(Fig. 26-26) provides another example of an NIH shift.
This reaction, which is catalyzed by the Fe(II)-containing
p-hydroxyphenylpyruvate dioxygenase, involves the oxida-
tive decarboxylation of an ␣-keto acid as well as ring hy-
droxylation. In this case, the NIH shift involves migration
of an alkyl group rather than of a hydride ion to form a
more stable carbocation (Fig. 26-31). This shift, which has
been demonstrated through isotope-labeling studies (rep-
resented by the different symbols in Figs. 26-26 and 26-31),
accounts for the observation that C3 is bonded to C4 in
p-hydroxyphenylpyruvate but to C5 in homogentisate.
d. Alkaptonuria and Phenylketonuria Result from
Defects in Phenylalanine Degradation
Archibald Garrod realized in the early 1900s that human
genetic diseases result from specific enzyme deficiencies.
We have repeatedly seen how this realization has con-
tributed to the elucidation of metabolic pathways. The first
such disease to be recognized was alkaptonuria, which,Gar-
rod observed, resulted in the excretion of large quantities of
homogentisic acid.This condition results from deficiency of
homogentisate dioxygenase (Fig. 26-26, Reaction 4).Alkap-
tonurics suffer no ill effects other than arthritis later in life
(although their urine darkens alarmingly because of the
rapid air oxidation of the homogentisate they excrete).
Individuals suffering from phenylketonuria (PKU) are
not so fortunate. Severe mental retardation occurs within a
few months of birth if the disease is not detected and treated
immediately (see below). Indeed, ⬃1% of the patients in
mental institutions were, at one time (before routine screen-
ing), phenylketonurics. PKU is caused by the inability to hy-
droxylate phenylalanine (Fig. 26-26, Reaction 1) and there-
fore results in increased blood levels of phenylalanine
(hyperphenylalaninemia). The excess phenylalanine is
transaminated to phenylpyruvate
by an otherwise minor pathway. The “spillover” of phen-
ylpyruvate (a phenylketone) into the urine was the first ob-
servation connected with the disease and gave the disease
its name, although it has since been demonstrated that it is
the high concentration of phenylalanine itself that gives rise
to brain dysfunction. All babies born in the United States
are now screened for PKU immediately after birth by test-
ing for elevated levels of phenylalanine in the blood.
Classic PKU results from a deficiency in phenylalanine
hydroxylase (PAH). When this was established in 1947, it
was the first human inborn error of metabolism whose ba-
sic biochemical defect had been identified. Since then, over
O
C
Phenylpyruvate
CH
2
COO
–
Section 26-3. Metabolic Breakdown of Individual Amino Acids 1045
Figure 26-29 The active site of the Fe(II) form of phenylalanine
hydroxylase (PAH) in complex with 5,6,7,8-tetrahydrobiopterin
(BH
4
). The Fe(II) (orange sphere) is octahedrally coordinated
(gray lines) by His 285, His 290, and Glu 330 (C green, N blue,
and O red) and three water molecules (red spheres). BH
4
atom
O4 is hydrogen bonded (black dashed lines) to two of these
water molecules. [Based on an X-ray structure by Edward
Hough, University of Tromsø, Norway. PDBid 1J8U.]
JWCL281_c26_1019-1087.qxd 6/8/10 9:38 AM Page 1045
400 mutations have been identified in PAH. Because all of
the tyrosine breakdown enzymes are normal, treatment
consists in providing the patient with a low-phenylalanine
diet and monitoring the blood level of phenylalanine to en-
sure that it remains within normal limits for the first 5 to 10
years of life (the adverse effects of hyperphenylalaninemia
seem to disappear after that age). PAH deficiency also ac-
counts for another common symptom of PKU: Its victims
have lighter hair and skin color than their siblings. This is
because tyrosine hydroxylation, the first reaction in the for-
mation of the black skin pigment melanin (Section 26-4B),
is inhibited by elevated phenylalanine levels.
Other causes of hyperphenylalaninemia have been discov-
ered since the introduction of infant screening techniques.
These result from deficiencies in the enzymes catalyzing the
formation or regeneration of 5,6,7,8-tetrahydrobiopterin
(BH
4
), the PAH cofactor (Fig. 26-28). In such cases, patients
must also be supplied with
L-3,4-dihydroxyphenylalanine
(
L-DOPA) and 5-hydroxytryptophan, metabolic precursors
of the neurotransmitters norepinephrine and serotonin, re-
spectively,since tyrosine hydroxylase and tryptophan hydrox-
ylase, the PAH homologs that produce these physiologically
active amines, also require 5,6,7,8-tetrahydrobiopterin (Sec-
tion 26-4B). Unfortunately, simply adding BH
4
to the diet of
1046 Chapter 26. Amino Acid Metabolism
hydrogen ion
migration
C
H
CH
3
OH
C
H
OH
R ⫽
Fe(IV) O
2–
Fe(II)
Pterin-4a-carbinolamine
O
H
2
N H
R
H
N
N
H
H
N
OH
H
N
1
6
5
2
O
H
2
N H
R
H
N
N
H
H
N
O
OH
Fe(II)
H
N
+
5,6,7,8-Tetrahydrobiopterin (BH
4
)
O
H
2
N
H
H
R
H
N
N
H
H
N
O
2
+
+
H
N
Fe(II)
Phenylalanine
C
NH
3
+
CO
2
–
H
H
2
C
3
H
3
4
H
+
3
4
H
+
C
NH
3
+
CO
2
–
H
H
2
C
3
H
+
HO
H
C
NH
3
+
CO
2
–
H
H
2
C
3
H
O
H
Tyrosine
HO
C
NH
3
+
CO
2
–
H
H
2
C
3
H
3
H
H
Resonance-stabilized oxonium ion
HO
C
NH
3
+
CO
2
–
H
H
2
C
+
3
H
+
H
HO
C
NH
3
+
CO
2
–
H
H
2
C
Figure 26-30 Proposed mechanism of the NIH shift in the phenylalanine hydroxylase
reaction. The mechanism involves (1 and 2) activation of oxygen by the enzyme’s BH
4
and Fe(II) cofactors to yield pterin-4a-carbinolamine and a reactive oxyferryl species
[Fe(IV)“O
2–
]; (3) reaction of the Fe(IV)“O
2–
with the phenylalanine substrate to form
an epoxide across its phenyl ring’s 3,4 bond; (4) epoxide opening to form a carbocation at
C3; (5) migration of a hydride from C4 to C3 to form a more stable carbocation (an
oxonium ion); and (6) ring aromatization to form tyrosine.
JWCL281_c26_1019-1087.qxd 4/20/10 9:26 AM Page 1046
an affected individual is not an effective treatment because
BH
4
is unstable and cannot cross the blood–brain barrier.
4 AMINO ACIDS AS
BIOSYNTHETIC PRECURSORS
Certain amino acids, in addition to their major function as
protein building blocks, are essential precursors of a variety
of important biomolecules, including nucleotides and nu-
cleotide coenzymes, heme, various hormones and neuro-
transmitters, and glutathione. In this section, we consider
the pathways producing some of these substances. We be-
gin by discussing the biosynthesis of heme from glycine and
succinyl-CoA. We then examine the pathways by which ty-
rosine, tryptophan, glutamate, and histidine are converted
to various neurotransmitters and study certain aspects of
glutathione biosynthesis and the involvement of this tripep-
tide in amino acid transport and other processes. Finally, we
consider the role of folate derivatives in the biosynthetic
transfer of C
1
units. The biosynthesis of nucleotides and
nucleotide coenzymes is the subject of Chapter 28.
A. Heme Biosynthesis and Degradation
Heme (Fig. 26-32), as we have seen, is an Fe-containing
prosthetic group that is an essential component of many
proteins, notably hemoglobin, myoglobin, and the cy-
tochromes. The initial reactions of heme biosynthesis are
common to the formation of other tetrapyrroles including
Section 26-4. Amino Acids as Biosynthetic Precursors 1047
Figure 26-31 The NIH shift in the p-hydroxyphenylpyruvate
dioxygenase reaction. Carbon atoms are labeled as an aid to
following the group migration constituting the shift.
Figure 26-32 Structure of heme. Heme’s C and N atoms are
derived from those of glycine and acetate.
O
–
–
O
CH
2
HO
CO
2
O
2
C
C
O
O
p-Hydroxyphenypyruvate
8
7
6
5
4
9
3
2
1
⌬
⌬
*
*
x
•
CH
2
HO
C
O
O
x
•
⌬
*
CH
2
HO
C
O
O
H
O
–
H
+
+
x
•
H
+
⌬
*
CH
2
OH
HO
C
O
H
O
–
x
•
+
⌬
OH
HO
H
*
CH
2
C
O
O
–
x
•
⌬
OH
HO
*
CH
2
C
O
O
–
x
•
+
⌬
OH
HO
H
*
CH
2
C
O
O
–
x
•
Resonance-stabilized
oxonium ion
Homogentisate
alkyl group
migration
COO
–
CH
3
COO
–
COO
–
COO
–
CH
2
CH
2
CH
3
CH
3
CH
2
CH
2
CH
2
CH
2
CH
2
H
3
C
CH
CH
CH
C
C
C
C
C
Fe
C
C
C
C
C
C
*
HC
*
C
*
C
H
C
H
C
N
*
N
*
N
*
N
*
C
*
H
3
N
H
3
C
C
+
*
*
Glycine
Acetate
A
D
C
B
+
␣
*
C
*
*
*
␥

␦
Heme
JWCL281_c26_1019-1087.qxd 4/20/10 9:26 AM Page 1047
chlorophyll in plants and bacteria (Section 24-2A) and
coenzyme B
12
in bacteria (Section 25-2Eb).
a. Porphyrins Are Derived from Succinyl-CoA
and Glycine
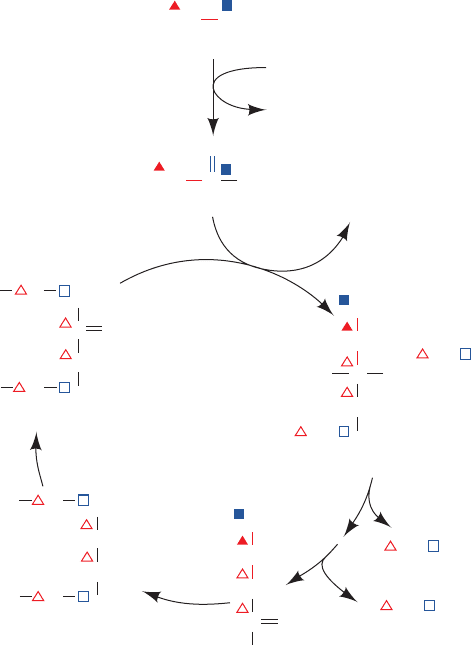
Elucidation of the heme biosynthesis pathway involved
some interesting detective work. David Shemin and David
Rittenberg, who were among the first to use isotopic trac-
ers in the elucidation of metabolic pathways, demon-
strated, in 1945, that all of heme’s C and N atoms can be de-
rived from acetate and glycine. Only glycine, out of a variety
of
15
N-labeled metabolites they tested (including ammo-
nia, glutamate, leucine, and proline), yielded
15
N-labeled
heme in the hemoglobin of experimental subjects to whom
these metabolites were administered. Similar experiments,
using acetate labeled with
14
C in its methyl or carboxyl
groups, or [
14
C
␣
]glycine, demonstrated that 24 of heme’s 34
carbon atoms are derived from acetate’s methyl carbon, 2
from acetate’s carboxyl carbon, and 8 from glycine’s C
␣
atom (Fig. 26-32). None of the heme atoms is derived from
glycine’s carboxyl carbon atom.
Figure 26-32 indicates that heme C atoms derived from
acetate methyl groups occur in groups of three linked
atoms. Evidently, acetate is first converted to some other
metabolite that has this labeling pattern. Shemin and Rit-
tenberg postulated that this metabolite is succinyl-CoA
based on the following reasoning (Fig. 26-33):
1. Acetate is metabolized via the citric acid cycle (Sec-
tion 21-1B).
2. Labeling studies indicate that atom C3 of the citric acid
cycle intermediate succinyl-CoA is derived from acetate’s
methyl C atom, whereas atom C4 comes from acetate’s
carboxyl C atom.
3. After many turns of the citric acid cycle, C1 and C2 of
succinyl-CoA likewise become fully derived from acetate’s
methyl C atom.
We shall see that this labeling pattern indeed leads to that
of heme.
In the mitochondria of yeast and animals as well as in
some bacteria, the first phase of heme biosynthesis is a con-
densation of succinyl-CoA with glycine followed by decar-
boxylation to form ␦-aminolevulinic acid (ALA) as catalyzed
by the PLP-dependent enzyme ␦-aminolevulinate synthase
(ALA synthase or ALAS; Fig. 26-34). The carboxyl group
lost in the decarboxylation (Fig. 26-34, Reaction 5) originates
in glycine, which is why heme contains no label from this
group.
b. The Pyrrole Ring Is the Product
of Two ALA Molecules
The pyrrole ring is formed in the next phase of the path-
way through linkage of two molecules of ALA to yield por-
phobilinogen (PBG). The reaction is catalyzed by porpho-
bilinogen synthase [PBGS; alternatively, ␦-aminolevulinic
acid dehydratase (ALAD)], which in yeast and mammals,
is Zn
2⫹
-dependent and involves Schiff base formation of
one of the substrate molecules with an enzyme amine
group (in some bacteria and all plants, Mg
2⫹
substitutes for
Zn
2⫹
). One possible mechanism of this condensation–
elimination reaction involves formation of a second Schiff
base between the ALA–enzyme Schiff base and the second
ALA molecule (Fig. 26-35). At this point, if we continue
tracing the acetate and glycine labels through the PBG syn-
thase reaction (Fig. 26-35), we can begin to see how heme’s
labeling pattern arises.
The X-ray structure of human PBGS in covalent com-
plex with its product PBG, determined by Jonathan
Cooper, indicates that this enzyme is a homooctamer with
D
4
symmetry. Each of its 330-residue subunits consists of
an ␣/ barrel and a 39-residue N-terminal tail that wraps
around a neighboring monomer (related to it by 2-fold
symmetry) so that the protein is better described as a rela-
tively loosely organized tetramer of compact dimers. As is
the case with nearly all ␣/ barrel enzymes, PBGS’s active
site (Fig. 26-36a) lies at the mouth of the barrel at the
1048 Chapter 26. Amino Acid Metabolism
Figure 26-33 The origin of the C atoms of succinyl-CoA as
derived from acetate via the citric acid cycle. C atoms labeled
with triangles and squares are derived, respectively, from
acetate’s methyl and carboxyl C atoms. Filled symbols label
atoms derived from acetate in the present round of the citric acid
cycle, whereas open symbols label atoms derived from acetate in
previous rounds of the citric acid cycle. Note that the C1 and C4
atoms of succinyl-CoA are scrambled on forming the 2-fold
symmetric succinate.
CoASH + ATP
AMP
+ PP
i
COO
–
CH
3
SCoA
CoASH
C
O
Acetate
CH
2
CH
2
CO
2
CO
2
COO
–
COO
–
CHO
COO
–
CH
2
CH
2
C
SCoA
O
COO
–
CH
3
Acetyl-CoA
Citric
acid
cycle
+
1
2
1
2
+
1
2
1
2
CH
2
CH
2
COO
–
COO
–
+
1
2
1
2
+
1
2
1
2
+
1
2
1
2
Citrate
Succinyl-CoA
Succinate
Oxaloacetate
+
1
2
1
2
CO
CH
2
COO
–
COO
–
+
1
2
1
2
+
1
2
1
2
JWCL281_c26_1019-1087.qxd 4/20/10 9:26 AM Page 1048
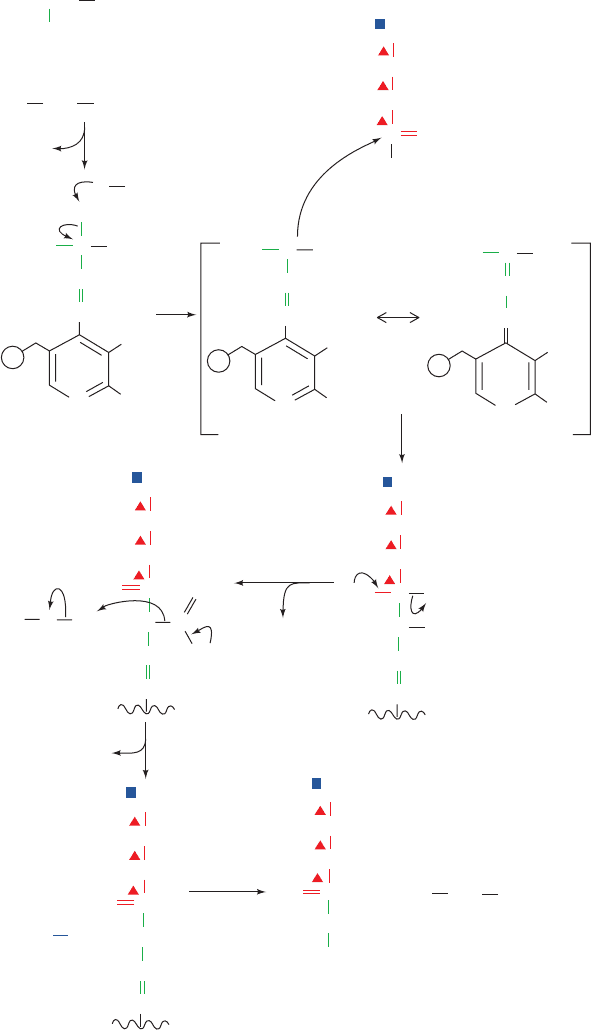
C-terminal ends of its  strands. The active site is covered
by a loop that comparison with other PBGS structures indi-
cates forms a flexible lid over the substrate, an arrange-
ment that is reminiscent of the glycolytic enzyme triose
phosphate isomerase (TIM; Fig. 17-11). PBG is covalently
bound to Lys 252 and its free amino group is coordinated to
the active site Zn
2⫹
ion. Lys 199 appears to be properly
positioned to act as a general acid–base catalyst.
Inhibition of PBG synthase by Pb
2⫹
(a competitor of its
active site Zn
2⫹
ion) is one of the major manifestations of
lead poisoning, which is among the most common acquired
environmental diseases. Indeed, it has been suggested that
the accumulation, in the blood, of ALA, which resembles
the neurotransmitter ␥-aminobutyric acid (Section 26-4B),
is responsible for the psychosis that often accompanies
lead poisoning.
Section 26-4. Amino Acids as Biosynthetic Precursors 1049
Figure 26-34 The mechanism of action of the PLP-dependent
enzyme ␦-aminolevulinate synthase (ALAS). The reaction steps
are (1) transimination, (2) PLP-stabilized carbanion formation,
6
4
3
2
1
H
+
Enz
B
B
5
+
␦-Aminolevulinate (ALA)
*
CH
2
CH
Enz
B
COO
–
CH
2
CO
2
C
O
C
O
CH
2
CH
HC
*
*
+
NH
*
+
NH
COO
–
O
–
CH
2
C
O
CH
2
*
CH
2
*
+
NH
3
COO
–
CH
2
C
O
CH
2
CoASH
SCoA
CH
HC
+
NH
*
*
COO
–
COO
–
CH
2
C
–
O
CH
2
CH
+
N
H
H
C
–
+
NH
*
*
COO
–
SCoA
COO
–
CH
2
CO
CH
2
P
CH
+
N
H
H
C
+
NH
*
CH
2
H
2
O
*
*
H
COO
–
COO
–
P
CH
N
H
HC
+
NH
*
*
COO
–
P
Succinyl-CoA
+
+
B
Enz
B
PLP
Enz
Glycine
*
NH
3
+
OH
OH
OH
CH
3
CH
3
CH
3
PLP
Enz
(3) C¬C bond formation, (4) CoA elimination, (5) decarboxylation
facilitated by the PLP–Schiff base, and (6) transimination
yielding ALA and regenerating the PLP–enzyme.
JWCL281_c26_1019-1087.qxd 4/20/10 9:26 AM Page 1049
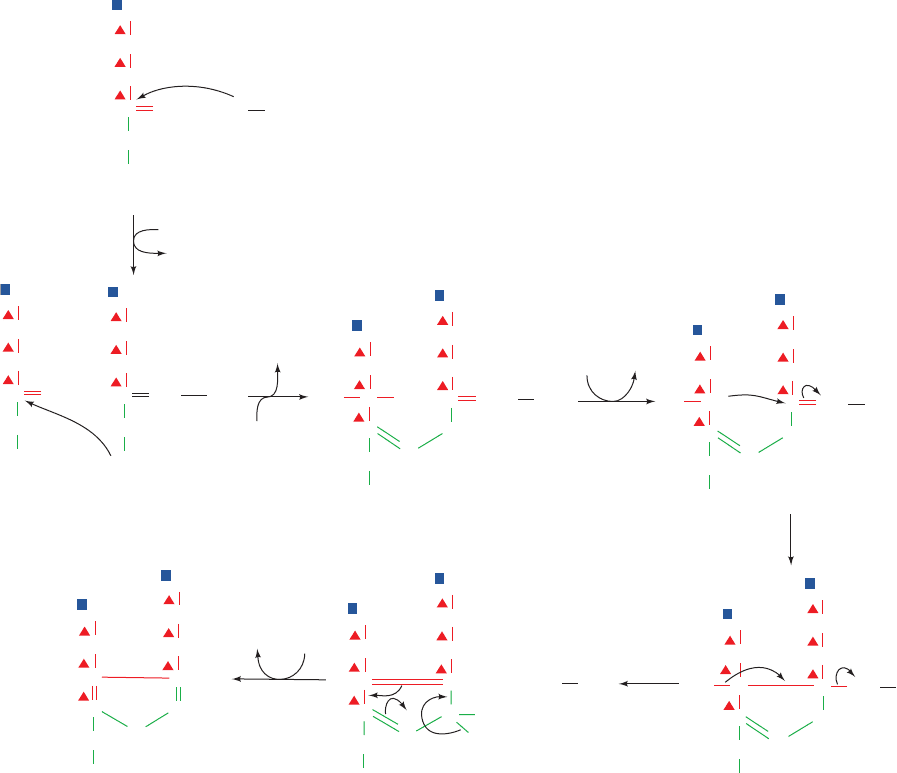
c. PBGS Has Two Quaternary States
with Different Enzymatic Activities
Although the X-ray structure of wild-type human PBGS
indicates that it is a D
4
-symmetric homooctamer, the X-ray
structure of its rare F12L mutant form, determined by
Eileen Jaffe, is a D
3
-symmetric homohexamer (Fig. 26-36b).
Moreover, the F12L mutant has far less activity than does
wild-type PBGS, even though residue 12 is far from the en-
zyme’s active site in both proteins.This is apparently due to
a conformational change in PBGS’s N-terminal arm: In the
octamer, the N-terminal arms of two adjacent monomers
wrap around each other’s barrel to form a so-called hug-
ging dimer, whereas in the hexamer, the N-terminal arms
extend away from the core of the protein to form a so-
called detached dimer (Fig. 26-36b). Nevertheless, the ␣/
barrels of these two oligomeric forms are closely superim-
posable. The difference in the activities of these two qua-
ternary forms is caused by the binding of an allosteric Mg
2⫹
ion in the octamer’s “hugging” interface that is absent in
the detached dimer.
The alternate oligomeric forms have been observed in
solutions of wild-type PBGS and the equilibrium between
these forms can be varied by changing the pH, the enzyme
concentration, and the substrate concentration, as well as
through mutation. Jaffe has therefore proposed that this
quaternary structural change is an allosteric mechanism for
controlling the activity of the enzyme. In contrast, in the
symmetry and sequential models of allosterism (Sections
10-4B and 10-4C), the quaternary state of the enzyme does
not change during an allosteric transition.
Jaffe has termed enzymes that control their activities by
changing their oligomeric states morpheeins [pronounced
morph-ee¿-in; derived from the verb “to morph” and the
classic pronunciation of the word protein (pro-tee¿-in)].Al-
though PBGS is the first known morpheein, several others
have since been identified (e.g., ribonucleotide reductase;
Section 28-3Ad). In fact, it may be that morpheeins are rel-
atively common because when an enzyme is purified, inac-
tive fractions are usually assumed to be denatured and are
therefore discarded rather than being characterized.
1050 Chapter 26. Amino Acid Metabolism
COO
–
CH
2
CH
2
H
2
N
*
CH
2
H
+
H
+
H
2
O
H
2
O
O
C
*
NH
2
*
CH
2
*
NH
2
COO
–
CH
2
CH
2
NH
C
ALA
+
Enz
Enz
*
CH
2
*
NH
2
COO
–
CH
2
CH
2
O
C
+
1
4
5
6
+
+
+
*
CH
2
*
CH
2
*
NH
2
H
*
N
COO
–
CH
2
C
C
H
H
Enz
COO
–
CH
2
CH
2
NH
+
BH
+
C
ALA
Porphobilinogen
(PBG)
2
+
*
CH
2
*
CH
2
*
NH
2
H
*
N
COO
–
CH
2
C
C
–
H
Enz
COO
–
CH
2
CH
2
NH
C
+
+
3
B
BH
+
B
*
CH
2
*
CH
2
*
NH
2
H
2
N
H
*
N
COO
–
CH
2
C
C
H
Enz
Enz
COO
–
CH
2
CH
2
NH
C
+
*
CH
2
*
C
H
H
*
NH
2
H
*
N
COO
–
CH
2
C
C
COO
–
CH
2
CH
2
C
+
*
CH
2
*
CH
*
NH
2
H
*
N
COO
–
CH
2
C
C
COO
–
CH
2
CH
2
C
Figure 26-35 A possible mechanism for porphobilinogen synthase. The reaction
involves (1) Schiff base formation, (2) second Schiff base formation, (3) formation
of a carbanion ␣ to a Schiff base, (4) cyclization by an aldol-type condensation,
(5) elimination of the enzyme¬NH
2
group, and (6) tautomerization.
JWCL281_c26_1019-1087.qxd 4/20/10 9:26 AM Page 1050