Voet D., Voet Ju.G. Biochemistry
Подождите немного. Документ загружается.

condensation of its aldehyde group with the ε-amino group
of an enzymatic Lys residue (Fig. 26-1d). This Schiff base,
which is conjugated to the coenzyme’s pyridinium ring, is
the focus of the coenzyme’s activity.
Esmond Snell,Alexander Braunstein, and David Metzler
demonstrated that the aminotransferase reaction occurs
via a Ping Pong Bi Bi mechanism whose two stages consist
of three steps each (Fig. 26-2):
Section 26-1. Amino Acid Deamination 1021
H
2
N
...
Enzyme
CH
2
()
4
H
C
O
–
CH
3
N
H
+
H
R C
H
COO
α
NH
2
..
–
+
...
Enzyme
CH
2
()
4
H
H
N
+
N
+
O
–
CH
3
N
H
R C
H
COO
α
N
..
–
CH
+
...
Enzyme
CH
2
()
4
H
NH
O
–
CH
3
N
H
R C
H
COO
α
–
CH
+
N
c
a
b
+
2
..
Enzyme–PLP
Schiff base
-Amino acid
␣
Geminal diamine
intermediate
•
Amino acid–PLP Schiff
base (aldimine)
...
H
O
–
CH
3
N
H
R
C COO
α
–
CH
+
N
+
Resonance-stabilized intermediate
–
...
H
O
–
CH
3
N
H
R
C COO
α
–
CH
..
N
+
EnzymeEnzyme
H
+
...
H
O
–
CH
3
N
H
R
C COO
α
–
CH
+
N
+
Enzyme
H
.
.
OH
–
.
.
H
2
N
Steps 2 & 2ⴕ: Tautomerization:
Ketimine
O
–
CH
3
N
H
R
C COO
α
–
+
NH
Carbinolamine
O
H
H C H
H
Steps 1 & 1ⴕ: Transimination:
Steps 3 & 3ⴕ: Hydrolysis:
Lys
.
.
H
2
N
.
.
H
2
N
H
2
NH
+
Lys Lys
EnzymeLys
O
–
CH
3
N
H
R
C COO
α
–
+
NH
Pyridoxamine
phosphate (PMP)–
enzyme
O
H C H
2
..
-Keto acid
α
+
EnzymeLys
•••
•
•
PO
3
O
2–
PO
3
O
2–
PO
3
O
2–
PO
3
O
2–
PO
3
O
2–
PO
3
O
2–
PO
3
O
2–
PO
3
O
2–
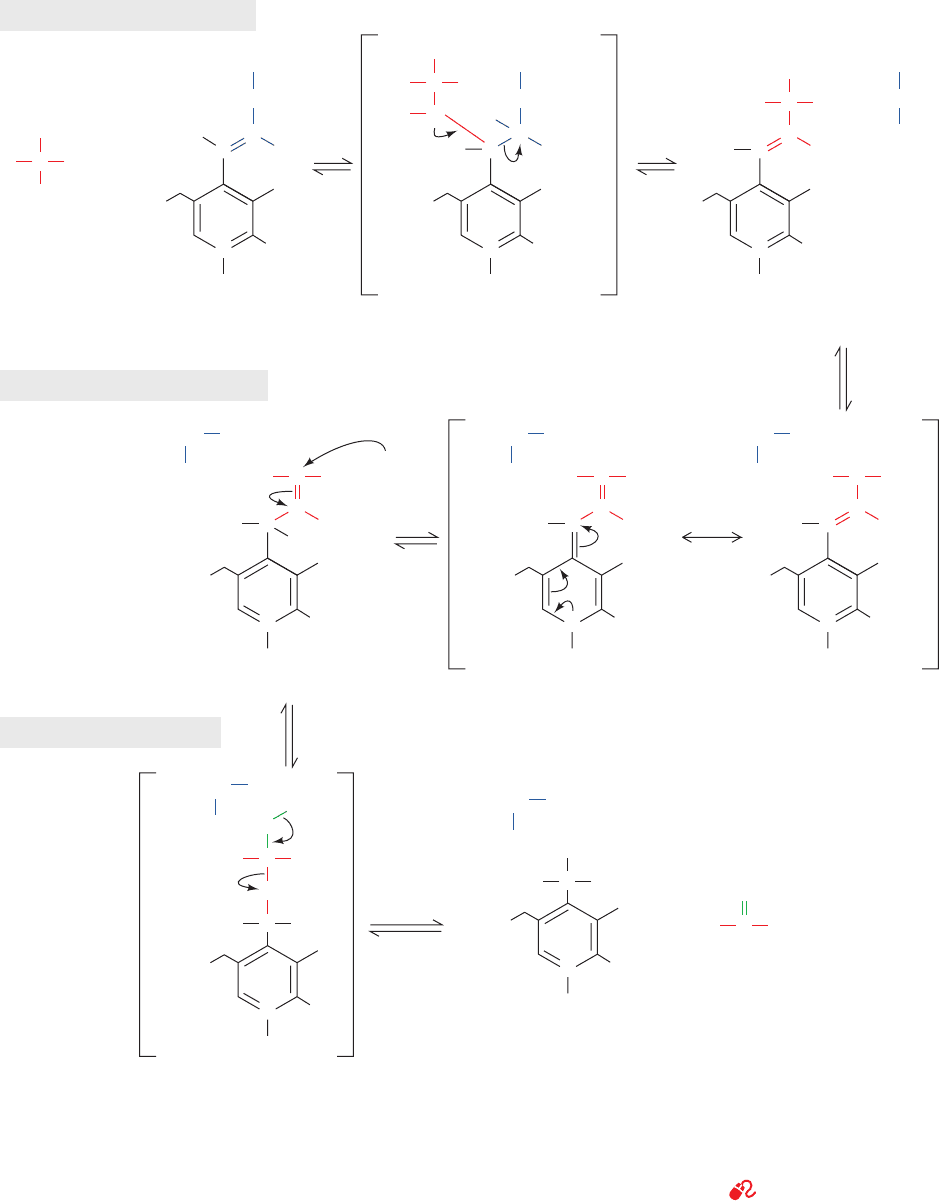
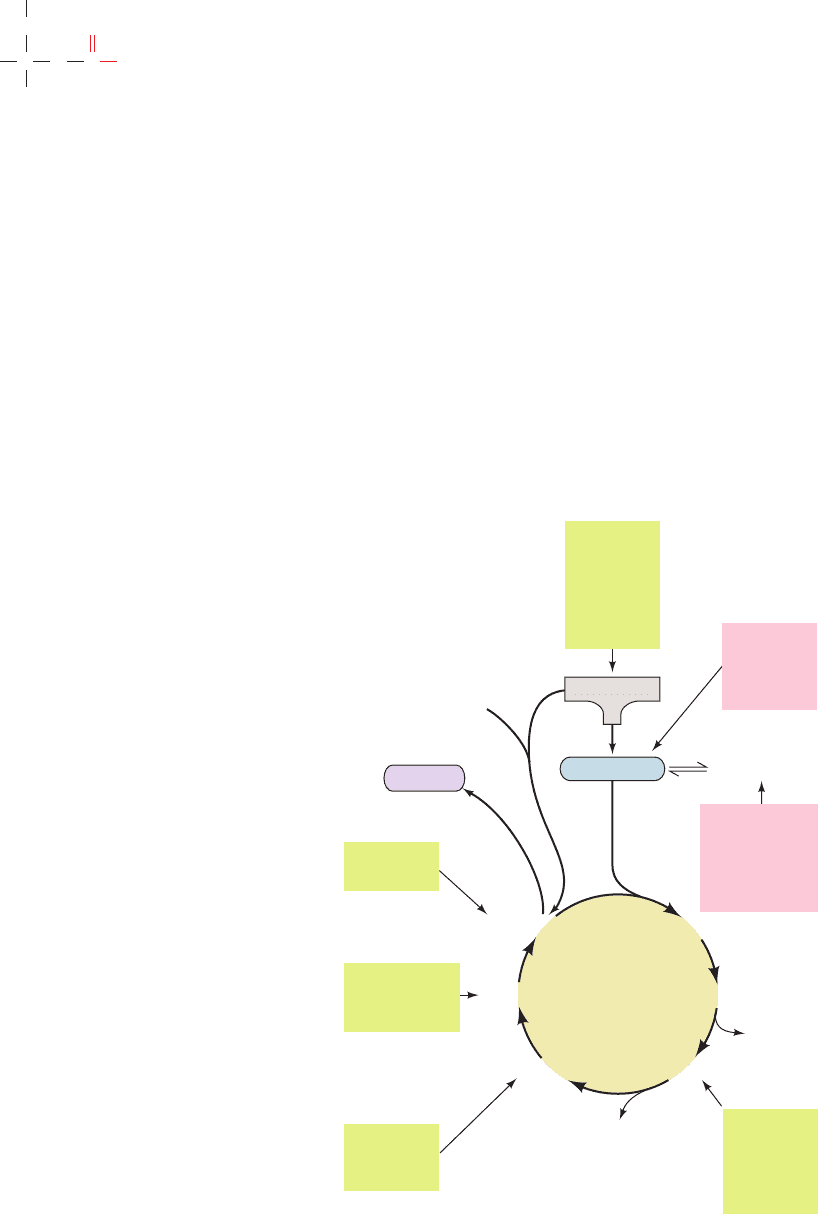
Figure 26-2 The mechanism of PLP-dependent enzyme-
catalyzed transamination. The first stage of the reaction, in which
the ␣-amino group of an amino acid is transferred to PLP
yielding an ␣-keto acid and PMP, consists of three steps:
(1) transimination; (2) tautomerization, in which the Lys released
during the transimination reaction acts as a general acid–base
catalyst; and (3) hydrolysis. The second stage of the reaction, in
which the amino group of PMP is transferred to a different
␣-keto acid to yield a new ␣-amino acid and PLP, is essentially
the reverse of the first stage: Steps 3¿,2¿, and 1¿ are, respectively,
the reverse of Steps 3, 2, and 1.
See the Animated Figures
JWCL281_c26_1019-1087.qxd 4/20/10 9:26 AM Page 1021
b. Stage I: Conversion of an Amino Acid
to an ␣-Keto Acid
Step 1. The amino acid’s nucleophilic amino group
attacks the enzyme–PLP Schiff base carbon atom in a tran-
simination (trans-Schiffization) reaction to form an amino
acid–PLP Schiff base (aldimine), with concomitant release
of the enzyme’s Lys amino group. This Lys is then free to
act as a general base at the active site.
Step 2. The amino acid–PLP Schiff base tautomerizes
to an ␣-keto acid–PMP Schiff base by the active site
Lys–catalyzed removal of the amino acid ␣ hydrogen and
protonation of PLP atom C4¿ via a resonance-stabilized
carbanion intermediate. This resonance stabilization facili-
tates the cleavage of the C
␣
¬H bond.
Step 3. The ␣-keto acid–PMP Schiff base is hydrolyzed
to PMP and an ␣-keto acid.
c. Stage II: Conversion of an ␣-Keto Acid
to an Amino Acid
To complete the aminotransferase’s catalytic cycle,
the coenzyme must be converted from PMP back to the
enzyme–PLP Schiff base. This involves the same three
steps as above, but in reverse order:
Step 3ⴕ. PMP reacts with an ␣-keto acid to form a
Schiff base.
Step 2ⴕ. The ␣-keto acid–PMP Schiff base tautomer-
izes to form an amino acid–PLP Schiff base.
Step 1ⴕ. The ε-amino group of the active site Lys
residue attacks the amino acid–PLP Schiff base in a trans-
imination reaction to regenerate the active enzyme–PLP
Schiff base, with release of the newly formed amino acid.
The reaction’s overall stoichiometry therefore is
Examination of the amino acid–PLP Schiff base’s struc-
ture (Fig. 26-2, Step 1) reveals why this system is called “an
electron-pusher’s delight.” Cleavage of any of the amino
acid C
␣
atom’s three bonds (labeled a, b, and c) produces a
resonance-stabilized C
␣
carbanion whose electrons are
␣-keto acid 1 ⫹ amino acid 2
Amino acid 1 ⫹␣-keto acid 2 Δ
delocalized all the way to the coenzyme’s protonated pyri-
dinium nitrogen atom; that is, PLP functions as an electron
sink. For transamination reactions, this electron-withdrawing
capacity facilitates removal of the ␣ proton (a bond cleav-
age) in the tautomerization of the Schiff base. PLP-
dependent reactions involving b bond cleavage (amino acid
decarboxylation) and c bond labilization are discussed in
Section 26-4B and in Sections 26-3Bb and 26-3G,respectively.
Aminotransferases differ in their specificity for amino
acid substrates in the first stage of the transamination reac-
tion, thereby producing the correspondingly different
␣-keto acid products. Most aminotransferases, however,
accept only ␣-ketoglutarate or (to a lesser extent) oxalo-
acetate as the ␣-keto acid substrate in the second stage of
the reaction, thereby yielding glutamate or aspartate as
their only amino acid products. The amino groups of most
amino acids are consequently funneled into the formation of
glutamate or aspartate, which are themselves interconverted
by glutamate–aspartate aminotransferase:
Oxidative deamination of glutamate (Section 26-1B) yields
ammonia and regenerates ␣-ketoglutarate for another
round of transamination reactions. Ammonia and aspartate
are the two amino group donors in the synthesis of urea.
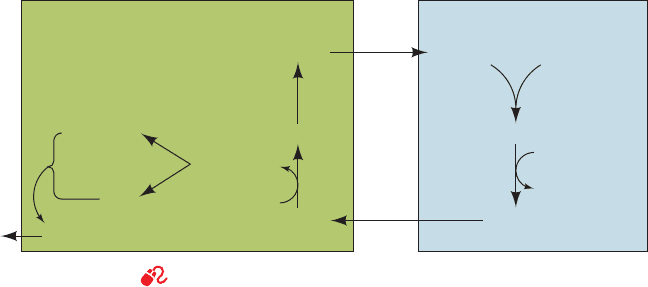
d. The Glucose–Alanine Cycle Transports Nitrogen
to the Liver
An important exception to the foregoing is a group of
muscle aminotransferases that accept pyruvate as their
␣-keto acid substrate. The product amino acid, alanine, is
released into the bloodstream and transported to the liver,
where it undergoes transamination to yield pyruvate for
use in gluconeogenesis (Section 23-1A).The resulting glu-
cose is returned to the muscles, where it is glycolytically
degraded to pyruvate. This is the glucose–alanine cycle
(Fig. 26-3). The amino group ends up in either ammonium
ion or aspartate for urea biosynthesis. Evidently, the
glucose–alanine cycle functions to transport nitrogen from
muscle to liver.
During starvation the glucose formed in the liver by this
route is also used by the other peripheral tissues, breaking the
␣-ketoglutarate ⫹ aspartate
Glutamate ⫹ oxaloacetate Δ
1022 Chapter 26. Amino Acid Metabolism
Figure 26-3 The glucose–alanine cycle. See the Animated Figures
Blood MuscleLiver
Glucose
AlanineAlanine
Glycogen
Pyruvate
Gluconeogenesis
Urea
Pyruvate
Glucose
α-Amino acid
α-Ketoacid
transamination
transamination
deamination
Glutamate
α-Ketoglutarate
Aspartate
transamination
NH
4
+
JWCL281_c26_1019-1087.qxd 6/21/10 10:39 AM Page 1022
cycle. Under these conditions both the amino group and the
pyruvate originate from muscle protein degradation, provid-
ing a pathway yielding glucose for other tissue use (recall that
muscle is not a gluconeogenic tissue; Section 23-1).
Nitrogen is also transported to the liver in the form of
glutamine, synthesized from glutamate and ammonia in a re-
action catalyzed by glutamine synthetase (Section 26-5Ab).
The ammonia is released for urea synthesis in liver mito-
chondria or for excretion in the kidney through the action
of glutaminase (Section 26-3D).
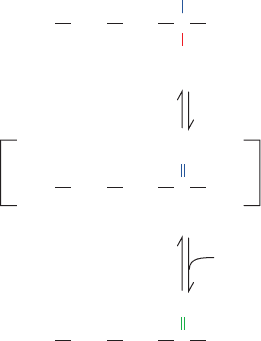
B. Oxidative Deamination: Glutamate
Dehydrogenase
Glutamate is oxidatively deaminated in the mitochondrial
matrix by glutamate dehydrogenase (GDH), the only
known enzyme that, in at least some organisms, can accept
either NAD
⫹
or NADP
⫹
as its redox coenzyme. Oxidation
is thought to occur with transfer of a hydride ion from gluta-
mate’s C
␣
to NAD(P)
⫹
, thereby forming ␣-iminoglutarate,
which is hydrolyzed to ␣-ketoglutarate and ammonia (Fig.
26-4). GDH is allosterically inhibited by GTP, NADH, and
nonpolar compounds such as palmitoyl-CoA and steroid
hormones. It is activated by ADP, NAD
⫹
, and leucine (the
most abundant amino acid in proteins;Table 4-1).
a. The X-Ray Structures of GDH Reveal its
Allosteric Mechanism
The X-ray structures of homohexameric GDH from
bovine and human liver mitochondria, determined by
Thomas Smith, reveal that each monomer has three domains,
a substrate domain, a coenzyme domain, and an antenna do-
main.The protein, which has D
3
symmetry, can be considered
to be a dimer of trimers, with the antenna domains of each
trimer wrapping around each other about the 3-fold axis
(Fig. 26-5a). Structural comparison of a 501-residue
monomer of the bovine GDH–glutamate–NADH–GTP
complex (Fig. 26-5b) with that of the 96% identical human
apoenzyme (no active site or regulatory ligands bound;
Fig. 26-5c) reveals that, on binding ligands, the coenzyme
binding domain rotates about the so-called pivot helix so as
to close the cleft between the coenzyme and substrate do-
mains. Simultaneously, the antenna domain twists in a way
that unwinds one turn of the antenna helix that is connected
to the pivot helix. Although the closed form is required for
catalysis, the open form favors the association and dissocia-
tion of substrates and products. In the open state, Arg 463
(human numbering) in the center of the pivot helix inter-
acts with the activator ADP (whose binding site in the
bovine complex is occupied by the ADP moiety of an
NADH; Fig. 26-5b), whereas in the closed state, the side
chain of His 454 hydrogen-bonds to the ␥-phosphate of the
inhibitor GTP. The GTP binding site is distorted and blocked
in the open state so that GTP binding favors the closed form
of the enzyme. This results in tight binding of substrates and
products and hence inhibition of the enzyme. ADP binding
favors the open form, allowing product dissociation, and
therefore activates the enzyme. Allosteric interactions ap-
pear to be communicated between subunits through the
interactions of the antenna domains. In fact, bacterial GDHs,
which lack allosteric regulation, differ from mammalian
GDHs mainly by the absence of antenna domains.
b. Hyperinsulinism/Hyperammonemia (HI/HA)
Is Caused by Uncontrolled GDH Activity
Charles Stanley has reported a new form of congenital
hyperinsulinism that is characterized by hypoglycemia and
hyperammonemia (HI/HA; hyperammonemia is elevated
levels of ammonia in the blood) and has shown that it is
caused by mutations in GDH at the N-terminal end of its
pivot helix in the GTP binding site or in the antenna do-
main near its joint with the pivot helix. The mutant en-
zymes have reduced sensitivity to GTP inhibition but re-
tain their ability to be activated by ADP. The GDH
mutants S448P, H454Y, and R463A, which were respec-
tively designed to affect the antenna region, the GTP bind-
ing site, and the ADP binding site (Fig. 26-5b), all have de-
creased sensitivity to GTP inhibition (Fig. 26-6), with
H454Y and S448P, which were previously known to be as-
sociated with HI/HA, conferring the most resistance to
GTP inhibition. The hypoglycemia and hyperammonemia
in HI/HA patients arises from the increased activity of the
GDH mutants in the breakdown direction, producing
increased amounts of ␣-ketoglutarate and NH
3
.The
increased levels of ␣-ketoglutarate stimulate the citric acid
cycle and oxidative phosphorylation, which has been
shown to lead to increased insulin secretion and hypo-
glycemia, thereby producing the symptoms of the disease.
The produced is usually converted to urea (Section
26-2) but can also be exported to the bloodstream.
If this scenario for the cause of HI/HA is correct, it
requires a reassessment of the role of GDH in ammonia
homeostasis.The equilibrium position of the GDH reaction
greatly favors the synthesis of Glu (⌬G°¿ ⬇ 30 kJ ⴢ mol
⫺1
for
the reaction as written in Fig. 26-4), but studies of cellular
NH
⫹
4
Section 26-1. Amino Acid Deamination 1023
Figure 26-4 The oxidative deamination of glutamate by
glutamate dehydrogenase. This reaction involves the
intermediate formation of ␣-iminoglutarate.
C COO
––
OOC CH
2
CH
2
NH
3
+
H
+ NAD(P)
+
Glutamate
C COO
––
OOC CH
2
CH
2
NH
2
+
+ NAD(P)
␣-Iminoglutarate
H + H
+
H
2
O
C COO
––
OOC CH
2
CH
2
NH
4
+
+
␣-Ketoglutarate
O
JWCL281_c26_1019-1087.qxd 4/20/10 9:26 AM Page 1023
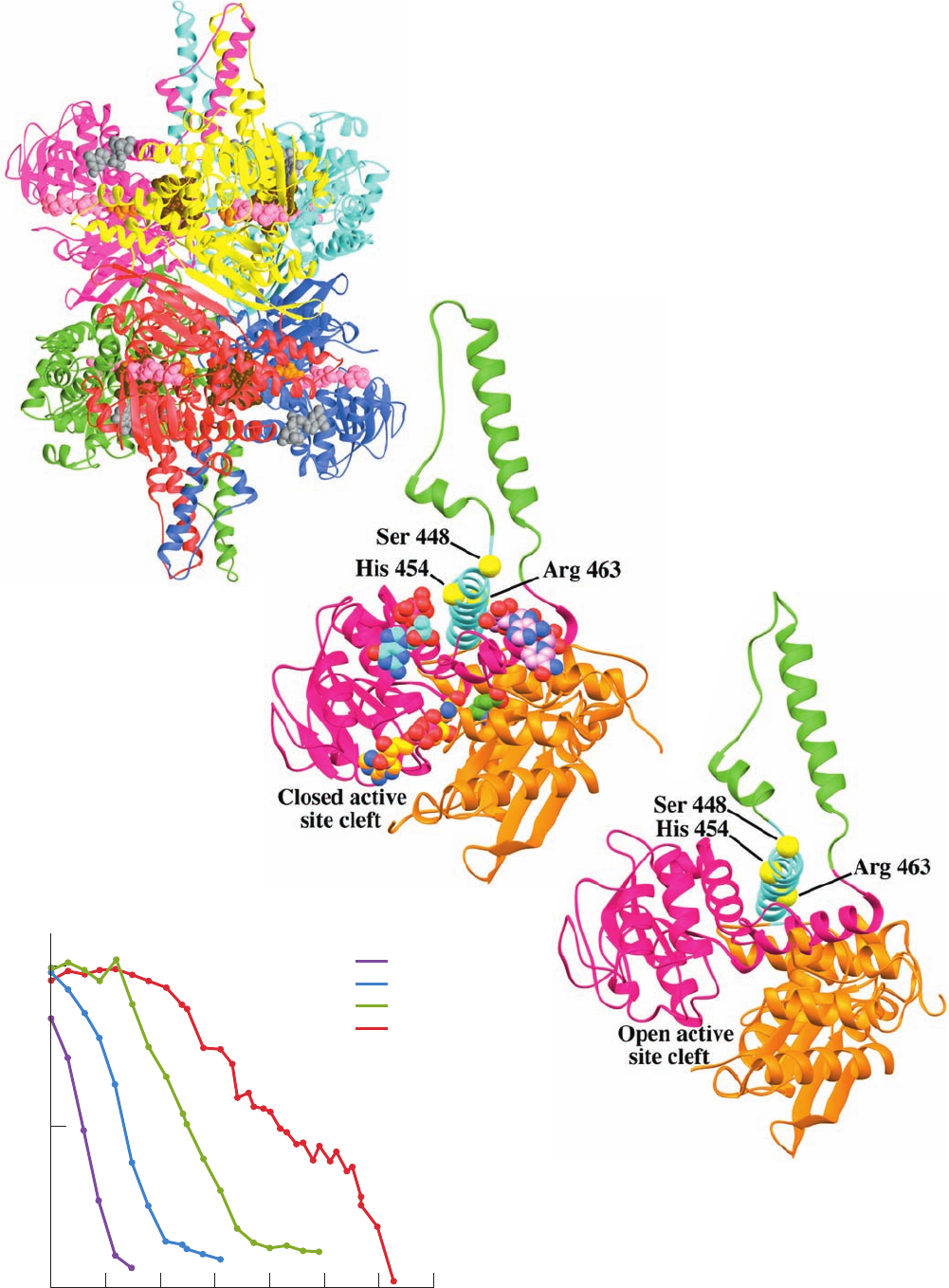
1024
Figure 26-6 Inhibition of human glutamate dehydrogenase
(GDH) by GTP. Human wild-type and mutant GDHs were
expressed in E. coli and assayed for sensitivity to GTP inhibition.
The midpoint of each curve corresponds to the concentration of
GTP causing 50% inhibition. [After Fang, J., Hsu, B.Y.L.,
MacMullen, C.M., Poncz, M., Smith,T.J., and Stanley, C.A.,
Biochem. J. 363, 81 (2002)].
0.01 0.1 1 10 10,0001000100 100,00
0
50
100
0
GDH activity (% basal)
GTP (μmol/L)
W/T
S448P
H454Y
R463A
Figure 26-5 X-ray structures of glutamate dehydrogenase (GDH).
(a) Bovine GDH in complex with glutamate, NADH, and GTP. The
homohexameric enzyme, which has D
3
symmetry, is viewed along one of its
2-fold axes with its 3-fold axis vertical. Each of its subunits is differently
colored.The bound substrates and ligands are shown in space-filling form
with glutamate orange, the substrate NADH pink, the NADH bound at the
ADP effector site brown, and the GTP effector gray. (b) One subunit of the
bovine GDH–glutamate–NADH–GTP complex drawn with the coenzyme
binding domain magenta, the substrate binding domain orange, the antenna
domain green, and the pivot helix cyan.The substrates and ligands are shown
in space-filling form colored according to atom type with glutamate C green,
substrate NADH C gold, ADP site-bound NADH C pink, GTP C cyan, N
blue, O red, and P magenta. The C
␣
atoms of Ser 448, His 454, and Arg 463
(human numbering) are represented by yellow spheres.
(c) One subunit of human apoGDH with the protein
colored as and viewed similarly to Part b. [Based on X-ray
structures by Thomas Smith, Donald Danforth Plant
Science Center, St. Louis, Missouri. PDBids (a and b)
1HWX and (c) 1L1F.]
(a)
(b)
(c)
JWCL281_c26_1019-1087.qxd 4/20/10 9:26 AM Page 1024
substrate and product concentrations suggested that the
enzyme functions close to equilibrium (⌬G ⬇ 0) in vivo. It
was therefore widely accepted that increases in [NH
3
], high
levels of which are toxic, would cause GDH to act in re-
verse, removing NH
3
and hence preventing its buildup to
toxic levels. However,since HI/HA patients have increased
GDH activity yet have higher levels of NH
3
than normal,
this accepted role of GDH cannot be correct. Indeed, if
GDH functioned close to equilibrium,changes in its activity
resulting from allosteric interactions would not result in
significant flux changes.
C. Other Deamination Mechanisms
Two nonspecific amino acid oxidases,
L-amino acid oxidase
and
D-amino acid oxidase, catalyze the oxidation of L- and
D-amino acids, utilizing FAD as their redox coenzyme
[rather than NAD(P)
⫹
].The resulting FADH
2
is reoxidized
by O
2
.
D-Amino acid oxidase occurs mainly in kidney. Its function
is an enigma since
D-amino acids are associated mostly
with bacterial cell walls (Section 11-3Ba). A few amino
acids, such as serine and histidine, are deaminated nonox-
idatively (Sections 26-3B and 26-3D).
2 THE UREA CYCLE
Living organisms excrete the excess nitrogen resulting
from the metabolic breakdown of amino acids in one of
three ways. Many aquatic animals simply excrete ammonia.
Where water is less plentiful, however, processes have
evolved that convert ammonia to less toxic waste products
that therefore require less water for excretion. One such
product is urea, which is excreted by most terrestrial verte-
brates; another is uric acid, which is excreted by birds and
terrestrial reptiles:
Accordingly, living organisms are classified as being either
ammonotelic (ammonia excreting), ureotelic (urea excret-
ing), or uricotelic (uric acid excreting). Some animals can
shift from ammonotelism to ureotelism or uricotelism if
their water supply becomes restricted. Here we focus our
attention on urea formation. Uric acid biosynthesis is dis-
cussed in Section 28-4A.
Urea is synthesized in the liver by the enzymes of the
urea cycle. It is then secreted into the bloodstream and
O
O
C
A
mmonia Urea
Uric acid
O
O
HN
N
H
N
H
H
N
H
2
NNH
3
NH
2
FADH
2
⫹ O
2
¡
FAD ⫹ H
2
O
2
␣-keto acid ⫹ NH
3
⫹ FADH
2
Amino acid ⫹ FAD ⫹ H
2
O
¡
sequestered by the kidneys for excretion in the urine. The
urea cycle was elucidated in outline in 1932 by Hans Krebs
and Kurt Henseleit (the first known metabolic cycle; Krebs
did not elucidate the citric acid cycle until 1937). Its indi-
vidual reactions were later described in detail by Sarah
Ratner and Philip Cohen.The overall urea cycle reaction is
Thus, the two urea nitrogen atoms are contributed by NH
3
and aspartate, whereas the carbon atom comes from
HCO
⫺
3
. Five enzymatic reactions are involved in the urea
cycle, two of which are mitochondrial and three cytosolic
(Fig. 26-7). In this section, we examine the mechanisms of
these reactions and their regulation.
A. Carbamoyl Phosphate Synthetase: Acquisition
of the First Urea Nitrogen Atom
Carbamoyl phosphate synthetase (CPS) is technically not
a urea cycle enzyme. It catalyzes the condensation and ac-
tivation of NH
3
and to form carbamoyl phosphate,
the first of the cycle’s two nitrogen-containing substrates,
with the concomitant hydrolysis of two ATPs. Eukaryotes
have two forms of CPS:
1. Mitochondrial CPS I uses NH
3
as its nitrogen donor
and participates in urea biosynthesis.
2. Cytosolic CPS II uses glutamine as its nitrogen donor
and is involved in pyrimidine biosynthesis (Section 28-2A).
The reaction catalyzed by CPS I involves three steps
(Fig. 26-8):
1. Activation of by ATP to form carboxyphos-
phate and ADP.
2. Nucleophilic attack of NH
3
on carboxyphosphate,
displacing the phosphate to form carbamate and P
i
.
3. Phosphorylation of carbamate by the second ATP to
form carbamoyl phosphate and ADP.
The reaction is essentially irreversible and is the rate-
limiting step of the urea cycle. CPS I is subject to allosteric
activation by N-acetylglutamate as is discussed in Section
26-2F.
E. coli contains only one type of CPS, which is homolo-
gous to both CPS I and CPS II.The enzyme is a heterodimer
but when allosterically activated by ornithine (a urea cycle
intemediate), it forms a tetramer of heterodimers, (␣)
4
. Its
small subunit (382 residues) functions to hydrolyze gluta-
mine and deliver the resulting NH
3
to its large subunit (1073
residues). However, if the enzyme’s glutaminase (glutamine
HCO
⫺
3
HCO
⫺
3
CH
Aspartate
NH
+
3
O
COO
–
CH
2
–
OOC
NH
3
+ HCO
–
3
+
Urea Fumarate
CH CH
NH
2
+ COO
––
OOC
C
H
2
N
3ATP
2ADP
+ 2P
i
+ AMP + PP
i
Section 26-2. The Urea Cycle 1025
JWCL281_c26_1019-1087.qxd 4/20/10 9:26 AM Page 1025
1026 Chapter 26. Amino Acid Metabolism
2ATP +
H
2
N NH
2
HCO
3
–
H
+
+
H
2
O
COO
–
NH
3
+
(
CH
2
)
3
CH NH
3
+
O
C
NH
2
+
C
H
2
N
NH
COO
–
(CH
2
)
3
CH NH
3
+
COO
–
CH
HC
COO
–
COO
–
CH
2
HC
COO
–
N
H
C
NH
(CH
2
)
3
COO
–
C NH
3
+
H
NH
2
+
COO
–
CH
2
HC
COO
–
NH
3
+
NH
(CH
2
)
3
COO
–
HC NH
3
+
OC
NH
2
+ NH
3
H
2
N
O
C
OPO
3
2–
+ 2ADP + P
i
CO
2
+ ATP
ADP + P
i
1
Ornithine
P
i
2
Citrulline
ATP
AMP + PP
i
3
Arginino-
succinate
Fumarate
4
Arginine
Urea
5
Cytosol
Mitochondrion
Carbamoyl phosphate
Aspartate
Ornithine
Urea
cycle
Citrulline
H
2
O
COO
–
CH
CH
2
COO
–
Malate
fumarase
OH
COO
–
C
CH
2
COO
–
Oxaloacetate
O
NADH
NAD
+
Gluconeogenesis
NAD(P)H
NAD(P)
+
malate dehydrogenase
Glutamate
Glutamate
transaminase
trans-
aminase
pyruvate
carboxylase
Amino acid
COO
–
NH
3
+
Alanine
CHH
3
C
␣-Keto acid
COO
–
CH
3
C
O
Pyruvate
Pyruvate
Oxaloacetate
Aspartate
␣-Ketoglutarate
␣-Ketoglutarate
␣-Ketoglutarate
glutamate
dehydrogenase
Glutamate
transaminase
+ H
+
JWCL281_c26_1019-1087.qxd 4/20/10 9:26 AM Page 1026
amidotransferase) activity is eliminated (e.g., by site-
directed mutagenesis), the large subunit can still produce
carbamoyl phosphate if NH
3
is supplied in high enough
concentration. The large subunit is composed of two nearly
superimposable halves that have 40% sequence identity.
The N-terminal half contains the carboxyphosphate syn-
thetic component and an oligomerization domain while the
C-terminal half contains the carbamoyl phosphate syn-
thetic component and an allosteric binding domain.
a. E. coli CPS Contains an Extraordinarily Long Tunnel
The X-ray structure of E. coli CPS in complex with
Mn
2⫹
,ADP, P
i
, and ornithine, determined by Hazel Holden
and Ivan Rayment, reveals that the active site for synthesis
of the carboxyphosphate intermediate is ⬃45 Å away from
the ammonia synthesis site and also ⬃35 Å away from the
carbamoyl phosphate synthesis active site. Astonishingly,
the three sites are connected by a narrow 96-Å-long molec-
ular tunnel that runs nearly the length of the elongated
protein molecule (Fig. 26-9). It therefore appears that CPS
guides its intermediate products from the active site in
which they are formed to that in which they are utilized.
This phenomenon, in which the intermediate of two reac-
tions is directly transferred from one enzyme active site to
Section 26-2. The Urea Cycle 1027
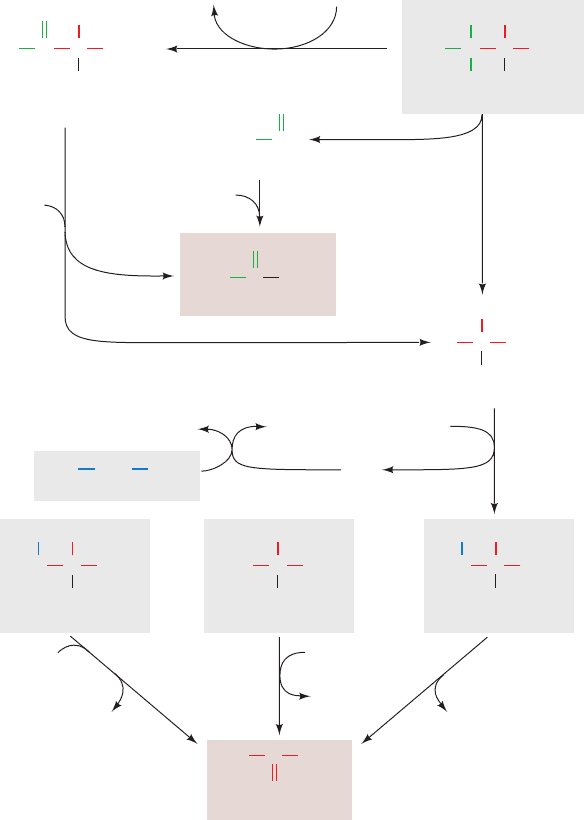
Figure 26-7 (Opposite) The urea cycle. Its five enzymes are
(1) carbamoyl phosphate synthetase, (2) ornithine
transcarbamoylase, (3) argininosuccinate synthetase,
(4) argininosuccinase, and (5) arginase. The reactions occur in
part in the mitochondrion and in part in the cytosol with
ornithine and citrulline being transported across the mitochondrial
membrane by specific transport systems (yellow circles). One of
the urea amino groups (green) originates as the NH
3
product of
the glutamate dehydrogenase reaction (top).The other amino
group (red) is obtained from aspartate through the transfer of an
amino acid to oxaloacetate via transamination (right).The
fumarate product of the argininosuccinase reaction is converted
to oxaloacetate for entry into gluconeogenesis via the same
reactions that occur in the citric acid cycle but take place in the
cytosol (bottom).The ATP utilized in Reactions 1 and 3 of the
cycle can be regenerated by oxidative phosphorylation from the
NAD(P)H produced in the glutamate dehydrogenase (top) and
malate dehydrogenase (bottom) reactions.
See the Animated
Figures
Figure 26-8 The mechanism of action of CPS I.
(1) Activation of HCO
⫺
3
by phosphorylation forms the
intermediate, carboxyphosphate; (2) nucleophilic attack on
carboxyphosphate by NH
3
forms the reaction’s second
intermediate, carbamate; and (3) phosphorylation of carbamate
by ATP yields the reaction product carbamoyl phosphate.
Figure 26-9 X-ray structure of E. coli carbamoyl phosphate
synthetase (CPS). The protein is represented by its C
␣
backbone.
The small subunit (magenta) contains the glutamine binding site
where NH
3
is produced or bound.The large subunit consists of
the carboxyphosphate domain (green), the oligomerization
domain (yellow), the carbamoyl phosphate domain (blue), and
the allosteric binding domain (orange).The 96-Å-long tunnel
connecting the three active sites is outlined in red. [Courtesy of
Hazel Holden and Ivan Rayment, University of Wisconsin.
PDBid 1JDB.]
1
ADP
O
O
–
ADP
2
3
P
i
ADP
Carboxyphosphate
PO
–
O
ADP
O
O
–
PO
–
O
C
O
O
–
HO +
Carbamate
ATP
NH
2
C
O
–
O
Carbamoyl phosphate
NH
2
2–
O
3
P C
O
O
OPO
2
3
–
C
O
–
O
:NH
3
+
JWCL281_c26_1019-1087.qxd 6/8/10 9:38 AM Page 1027
another, is called channeling (the term “tunneling” is re-
served for certain quantum mechanical phenomena).
Channeling increases the rate of a metabolic pathway by
preventing the loss of its intermediate products as well as
protecting the intermediate from degradation. NH
3
must
travel ⬃45 Å down the CPS tunnel to react with car-
boxyphosphate to form the next intermediate, carbamate.
The carbamate, in turn, must travel an additional ⬃35 Å to
the site where it is phosphorylated by ATP to form the final
product carbamoyl phosphate. The NH
3
transfer tunnel is
lined with polar groups capable of forming hydrogen bonds
with NH
3
, whereas the tunnel through which carbamate trav-
els is lined with backbone atoms and lacks charged groups
that might induce its hydrolysis as it diffuses between active
sites. Shielding and channeling are necessary because the in-
termediates carboxyphosphate and carbamate are extremely
reactive, having half-lives of 28 and 70 ms, respectively, at
neutral pH. Also, channeling allows the local concentration
of NH
3
to reach a higher value than is present in the cellular
medium.We shall encounter several other examples of chan-
neling in our studies of metabolic enzymes, but the CPS tun-
nel is far longer than that in any other known enzyme.
B. Ornithine Transcarbamoylase
Ornithine transcarbamoylase transfers the carbamoyl group
of carbamoyl phosphate to ornithine, yielding citrulline (Fig.
26-7, Reaction 2; note that both of these compounds are
“nonstandard” ␣-amino acids in that they do not occur in
proteins). The reaction occurs in the mitochondrion so that
ornithine, which is produced in the cytosol, must enter the
mitochondrion via a specific transport system. Likewise,
since the remaining urea cycle reactions occur in the cytosol,
citrulline must be exported from the mitochondrion.
C. Argininosuccinate Synthetase: Acquisition
of the Second Urea Nitrogen Atom
Urea’s second nitrogen atom is introduced in the urea cy-
cle’s third reaction by the condensation of citrulline’s urei-
do group with an aspartate amino group by argininosucci-
nate synthetase (Fig. 26-10). The ureido oxygen atom is
activated as a leaving group through formation of a
citrullyl–AMP intermediate, which is subsequently displaced
by the aspartate amino group. Support for the existence of
the citrullyl–AMP intermediate comes from experiments us-
ing
18
O-labeled citrulline (* in Fig. 26-10). The label was iso-
lated in the AMP produced by the reaction, demonstrating
that at some stage of the reaction, AMP and citrulline are
linked covalently through the ureido oxygen atom.
D. Argininosuccinase
With formation of argininosuccinate, all of the urea mole-
cule components have been assembled. However, the
amino group donated by aspartate is still attached to the
aspartate carbon skeleton.This situation is remedied by the
argininosuccinase-catalyzed elimination of arginine from
the aspartate carbon skeleton forming fumarate (Fig. 26-7,
Reaction 4). Arginine is urea’s immediate precursor. The
fumarate produced in the argininosuccinase reaction reacts
via the fumarase and malate dehydrogenase reactions to
form oxaloacetate (Fig. 26-7, bottom), which is then used in
gluconeogenesis (Section 23-1).
E. Arginase
The urea cycle’s fifth and final reaction is the arginase-
catalyzed hydrolysis of arginine to yield urea and regener-
ate ornithine (Fig. 26-7, Reaction 5). Ornithine is then re-
turned to the mitochondrion for another round of the cycle.
The urea cycle thereby converts two amino groups,one from
NH
3
and one from aspartate, and a carbon atom from
to the relatively nontoxic excretion product urea at
the cost of four “high-energy” phosphate bonds (three ATP
hydrolyzed to two ADP, two P
i
, AMP, and PP
i
, followed by
rapid PP
i
hydrolysis). This energetic cost, together with that
of gluconeogenesis, is supplied by the oxidation of the
acetyl-CoA formed by the breakdown of amino acid carbon
skeletons (e.g., threonine, Fig. 26-12). Indeed, half the oxy-
gen that the liver consumes is used to provide this energy.
F. Regulation of the Urea Cycle
Carbamoyl phosphate synthetase I, the mitochondrial en-
zyme that catalyzes the first committed reaction of the urea
HCO
⫺
3
1028 Chapter 26. Amino Acid Metabolism
Figure 26-10 The mechanism of action of argininosuccinate
synthetase. The steps involved are (1) activation of the ureido
oxygen of citrulline through the formation of citrullyl–AMP and
P AMP
+
O
**
ATP
Citrulline
C H
2
NC
COO
–
CH
2
COO
–
H
NH C
COO
–
CH
2
COO
–
H
O C
NH
2
CNH
+
3
H
COO
–
NH
2
NH
(CH
2
)
3
Citrullyl–AMP
Aspartate
CNH
+
3
H
COO
–
NH
(CH
2
)
3
P
A
MP
¨
¨
1
PP
i
2
AMP
*
+
C
NH
2
Argininosuccinate
CNH
+
3
H
COO
–
NH
(CH
2
)
3
+
+
(2) displacement of AMP by the ␣-amino group of aspartate. The
asterisk (*) traces the fate of
18
O originating in citrulline’s ureido
group.
JWCL281_c26_1019-1087.qxd 6/8/10 9:38 AM Page 1028
CO
2
Alanine
Cysteine
Glycine
Serine
Threonine
Tryptophan
Isoleucine
Leucine
Lysine
Threonine
Pyruvate
Acetyl-CoA Acetoacetate
Leucine
Lysine
Phenylalanine
Tryptophan
Tyrosine
Citric
acid
cycle
Citrate
Isocitrate
Oxaloacetate
Fumarate
Succinyl-CoA α
-Ketoglutarate
Glucose
CO
2
Asparagine
Aspartate
Aspartate
Phenylalanine
Tyrosine
Isoleucine
Methionine
Valine
CO
2
Arginine
Glutamate
Glutamine
Histidine
Proline
cycle, is allosterically activated by N-acetylglutamate:
This metabolite is synthesized from glutamate and acetyl-
CoA by N-acetylglutamate synthase and hydrolyzed by a
specific hydrolase.The rate of urea production by the liver is,
in fact,correlated with the N-acetylglutamate concentration.
Increased urea synthesis is required when amino acid break-
down rates increase, generating excess nitrogen that must be
excreted. Increases in these breakdown rates are signaled by
an increase in glutamate concentration through transamina-
tion reactions (Section 26-1). This situation, in turn, causes
an increase in N-acetylglutamate synthesis, stimulating car-
bamoyl phosphate synthetase and thus the entire urea cycle.
The remaining enzymes of the urea cycle are controlled
by the concentrations of their substrates.Thus, inherited de-
ficiencies in urea cycle enzymes other than arginase do not
result in significant decreases in urea production (the total
lack of any urea cycle enzyme results in death shortly after
birth). Rather, the deficient enzyme’s substrate builds up,
increasing the rate of the deficient reaction to normal. The
anomalous substrate buildup is not without cost, however.
The substrate concentrations become elevated all the way
back up the cycle to NH
3
, resulting in hyperammonemia.
Although the root cause of NH
3
toxicity is not completely
understood, high [NH
3
] puts an enormous strain on the
NH
3
-clearing system, especially in the brain (symptoms of
urea cycle enzyme deficiencies include mental retardation
and lethargy). This clearing system has been proposed to
involve glutamate dehydrogenase (working in reverse) and
glutamine synthetase, which decrease the ␣-ketoglutarate
and glutamate pools (Sections 26-1 and 26-5Ab).The brain
is most sensitive to the depletion of these pools. Depletion of
␣-ketoglutarate decreases the rate of the energy-generating
citric acid cycle, whereas decreasing the glutamate concen-
tration disturbs neuronal function, since it is both a neuro-
transmitter and a precursor to ␥-aminobutyrate (GABA),
another neurotransmitter (Section 20-5Cf). Glutamate de-
pletion would also decrease the functioning of the urea cy-
cle, since it is also the precursor to N-acetylglutamate, the
major regulator of the cycle. The involvement of GDH in
NH
3
clearance is a subject of debate in light of the observa-
tion that HI/HA involves deinhibition of GDH (Section
26-1Bb), suggesting that increased GDH activity increases
the NH
3
concentration rather than decreasing it.
3 METABOLIC BREAKDOWN
OF INDIVIDUAL AMINO ACIDS
The degradation of amino acids converts them to citric acid
cycle intermediates or their precursors so that they can be
metabolized to CO
2
and H
2
O or used in gluconeogenesis.
N-Acetylglutamate
C CCH
3
O
N
H
H
–
OOC
COO
–
(CH
2
)
2
Indeed, oxidative breakdown of amino acids typically ac-
counts for 10 to 15% of the metabolic energy generated by
animals. In this section we consider how amino acid carbon
skeletons are catabolized. The 20 “standard” amino acids
(the amino acids of proteins) have widely differing carbon
skeletons, so their conversions to citric acid cycle intermedi-
ates follow correspondingly diverse pathways. We shall not
describe all of the many reactions involved in detail.Rather,
we shall consider how these pathways are organized and fo-
cus on a few reactions of chemical and/or medical interest.
A. Amino Acids Can Be Glucogenic,
Ketogenic, or Both
“Standard” amino acids are degraded to one of seven meta-
bolic intermediates: pyruvate, ␣-ketoglutarate, succinyl-
CoA, fumarate, oxaloacetate, acetyl-CoA, or acetoacetate
(Fig. 26-11).The amino acids may therefore be divided into
two groups based on their catabolic pathways (Fig. 26-11):
1. Glucogenic amino acids, whose carbon skeletons are
degraded to pyruvate, ␣-ketoglutarate, succinyl-CoA,
fumarate, or oxaloacetate and are therefore glucose pre-
cursors (Section 23-1A).
Section 26-3. Metabolic Breakdown of Individual Amino Acids 1029
Figure 26-11 Degradation of amino acids to one of seven
common metabolic intermediates. Glucogenic and ketogenic
degradations are indicated in green and red, respectively.
JWCL281_c26_1019-1087.qxd 4/20/10 9:26 AM Page 1029
COO
H
3
C
C
Pyruvate
H
3
C
C
COO
–
NH
3
+
H
Alanine
O
H
3
C
CH
Acetaldehyde
O
C
H
3
C
C COO
–
H NH
3
+
HO H
Threonine
H
2
C
C COO
–
NH
3
+
HS H
Cysteine
H
3
C
C
Acetyl-CoA
O
SCoA
–
H
2
C
C COO
–
NH
3
+
HO H
Serine
H C COO
–
NH
3
+
H
Glycine
N
5
,N
10
-Methylene-THF
THF
NADH + NH
4
+
+ CO
2
NH
3
+
NAD
+
+ CH
2
Glycine
COO
C
H
3
C
C COO
–
NADH + H
+
CoA
NAD
+
NH
3
+
O
H
␣-Amino--ketobutyrate
–
α-Ketoglutarate
Glutamate
NH
3
SO
3
2–
H
2
S, , SCN
–
()
NH
3
+
1
2
34
5
6
7
several
paths
H
2
O
CoA
or
2. Ketogenic amino acids, whose carbon skeletons are
broken down to acetyl-CoA or acetoacetate and can thus
be converted to ketone bodies or fatty acids (Sections 25-3
and 25-4).
For example, alanine is glucogenic because its transamina-
tion product, pyruvate (Section 26-1A), can be converted
to glucose via gluconeogenesis (Section 23-1A). Leucine,
on the other hand, is ketogenic; its carbon skeleton is con-
verted to acetyl-CoA and acetoacetate (Section 26-3F).
Since animals lack any metabolic pathway for the net con-
version of acetyl-CoA or acetoacetate to gluconeogenic
precursors, no net synthesis of carbohydrates is possible
from leucine, or from lysine, the only other purely keto-
genic amino acid. Isoleucine, phenylalanine, threonine,
tryptophan, and tyrosine,however,are both glucogenic and
ketogenic; isoleucine, for example, is broken down to
succinyl-CoA and acetyl-CoA and hence is a precursor of
both carbohydrates and ketone bodies (Section 26-3Ed).
The remaining 13 amino acids are purely glucogenic.
In studying the specific pathways of amino acid break-
down, we shall organize the amino acids into groups that
are degraded into each of the seven metabolic intermedi-
ates mentioned above: pyruvate, oxaloacetate, ␣-ketoglu-
tarate, succinyl-CoA, fumarate, acetyl-CoA, and acetoac-
etate. When acetoacetyl-CoA is a product in amino acid
degradation, it can, of course, be directly converted to
acetyl-CoA (Section 25-3).We also discuss the pathway by
which, in liver, it is converted instead to acetoacetate for
use as an alternative fuel source in peripheral tissues (Sec-
tion 25-3).
B. Alanine, Cysteine, Glycine, Serine, and
Threonine Are Degraded to Pyruvate
Five amino acids, alanine, cysteine, glycine, serine, and threo-
nine, are broken down to yield pyruvate (Fig. 26-12).Trypto-
phan should also be included in this group since one of its
breakdown products is alanine (Section 26-3G),which,as we
have seen (Section 26-1Ad), is transaminated to pyruvate.
1030 Chapter 26. Amino Acid Metabolism
Figure 26-12 The pathways converting
alanine, cysteine, glycine, serine, and threonine to
pyruvate. The enzymes involved are (1) alanine
aminotransferase, (2) serine dehydratase,
(3) glycine cleavage system, (4) and (5) serine
hydroxymethyltransferase, (6) threonine
dehydrogenase, and (7) ␣-amino--ketobutyrate
lyase.
JWCL281_c26_1019-1087.qxd 4/20/10 9:26 AM Page 1030