Voet D., Voet Ju.G. Biochemistry
Подождите немного. Документ загружается.

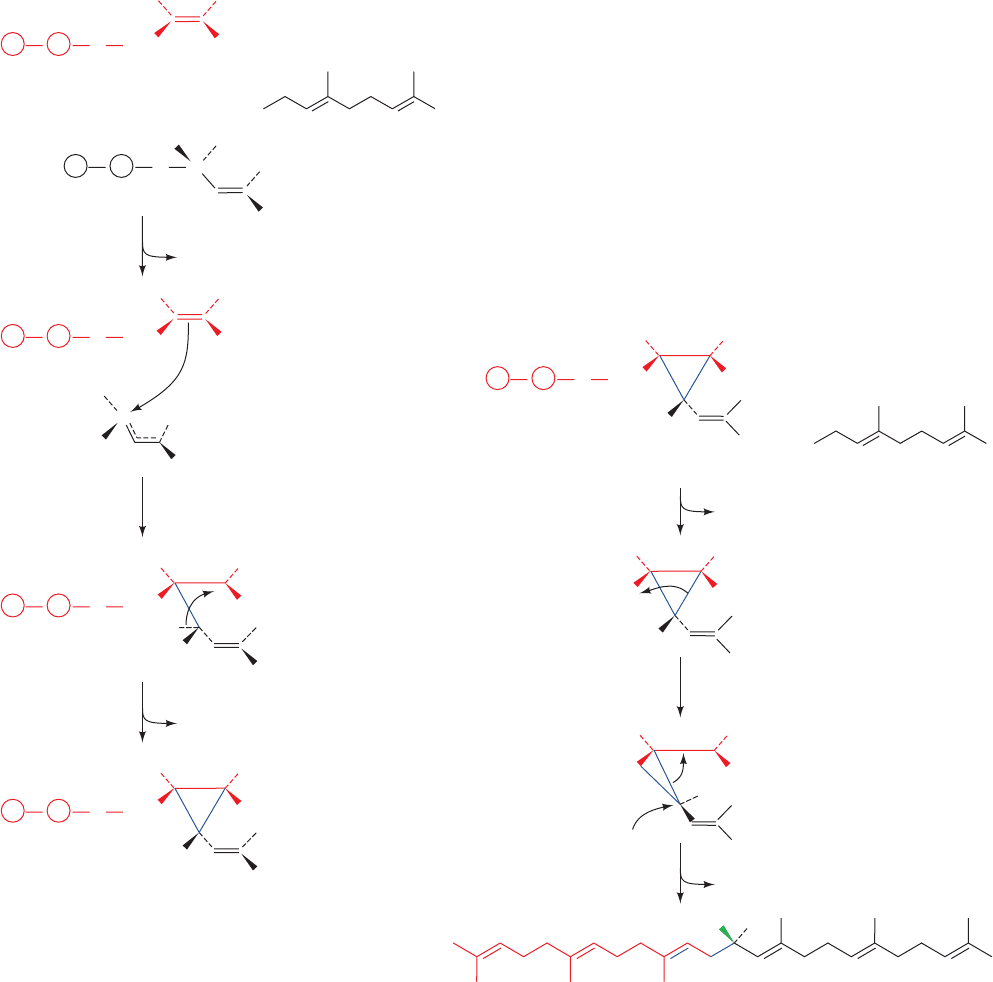
tion is initiated by the elimination of PP
i
from one farnesyl
pyrophosphate molecule to form an allylic carbocation at
C1 that is stabilized by a interaction with an essential Tyr
residue (Fig. 25-53). The highly reactive electron-deficient
carbocation inserts into the electron-rich C2“C3 double
bond of the second molecule, yielding presqualene py-
rophosphate, a cyclopropylcarbinyl pyrophosphate.
Step II The rearrangement and reduction of presqua-
lene pyrophosphate by NADPH to form squalene.This re-
action involves the formation and rearrangement of a cy-
clopropylcarbinyl cation in a complex reaction sequence
called a 1–2–3 process (Fig. 25-54).
SQS, a monomeric protein, is anchored to the ER mem-
brane via a short C-terminal transmembrane domain, with
its active site facing the cytosol. This allows it to accept its
water-soluble substrates, farnesyl pyrophosphate and
NADPH, from the cytosol and release its hydrophobic
product, squalene, in the ER membrane.
Section 25-6. Cholesterol Metabolism 981
H
1
1
23
HR
R
CH
3
CH
2
OPP
Presqualene pyrophosphate
3
H
H
1
1
1
23
HR
R
CH
2
CH
3
CH
3
CH
3
OPP
H
+
+
1
1
PP
i
2
1
H
CH
2
OPP
1
23
HR
CH
3
CH
2
23
R
CH
3
OPP
H
H
CH
3
R
C
OPP
H
C
R
H
CH
3
+
+
+
Tertiary
carbocation
Allylic
carbocation
Farnesyl
pyrophosphate
Farnesyl
pyrophosphate
R
=
Figure 25-53 Proposed mechanism for the formation of
presqualene pyrophosphate from two farnesyl pyrophosphate
molecules by squalene synthase (Fig. 25-52, Step I). (1) The
pyrophosphate group on one farnesyl pyrophosphate leaves,
yielding an allylic carbocation.This reaction step is facilitated by
proton donation from the side chain of an essential Tyr residue,
which then stabilizes the allylic cation via –cation interactions.
(2) The C2“C3 double bond of the second farnesyl
pyrophosphate nucleophilically attacks the allylic carbocation
to form a tertiary carbocation at C3. (3) The abstraction of the
pro-S proton at C1¿ by the phenolate group of the essential Tyr
residue results in the formation of a C1¿¬C3 bond yielding
presqualene pyrophosphate.
1
2
3
1
H
1
1
23
HR
RR
CH
3
CH
3
H
2
C
OPP
Squalene
PP
i
1
2
H
1
1
23
HR
R
CH
3
CH
3
H
2
C
=
Presqualene pyrophosphate
Primary
cyclopropylcarbinyl
cation
3
H
1
1
23
H
H H
R
R
CH
3
CH
3
Tertiary
cyclopropylcarbinyl
cation
+
+
+
NADPH
NADP
Figure 25-54 Mechanism of rearrangement and reduction of
presqualene pyrophosphate to squalene as catalyzed by squalene
synthase (Fig. 25-52, Step II). (1) Presqualene’s pyrophosphate
group leaves, yielding a primary carbocation at C1. (2) The
electrons forming the C1¿¬C3 bond migrate to C1, forming
squalene’s C1¬C1¿ bond and a tertiary carbocation at C3. (3)
The process is completed by the addition of an NADPH-supplied
hydride ion to C1¿ and the formation of the C2“C3 double
bond.
JWCL281_c25_940-1018.qxd 6/8/10 8:59 AM Page 981

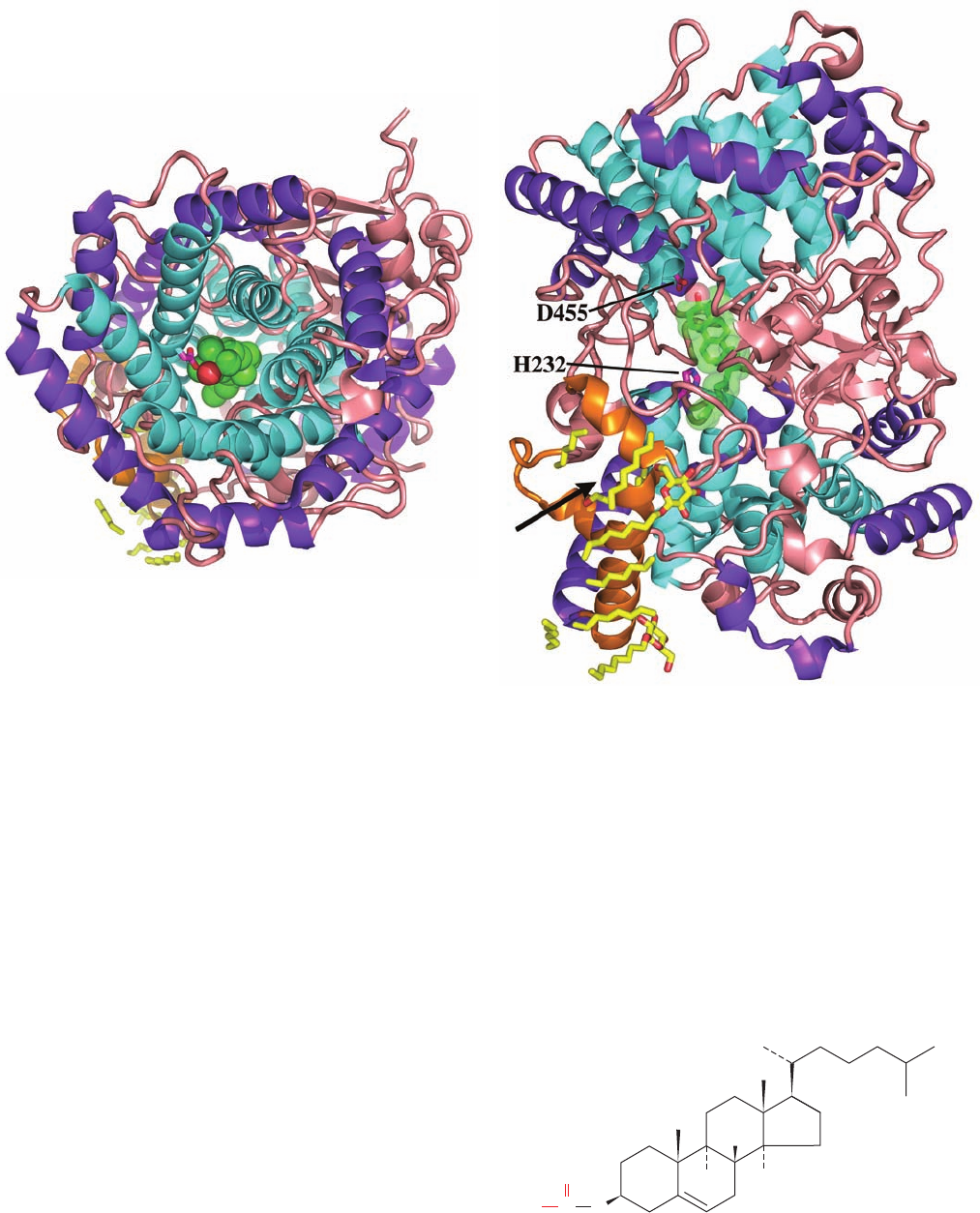
The X-ray structure of the 417-residue human SQS, in
complex with the inhibitor CP-320473,
determined by Jayvardhan Pandit, reveals that the protein
folds as a single domain with a large channel across one
face into which CP-320473 binds (Fig. 25-55). The channel
is lined with Asp and Arg residues that mutagenesis studies
indicate are involved in FPP binding. Of these, the con-
served Asp 80 and Asp 84 are implicated in binding Mg
2
ions that ligand an FPP pyrophosphate group. These Asp
residues are adjacent to Tyr 171, which forms the base of
the channel and which mutagenesis studies have identified
as the essential Tyr that is implicated in stabilizing the al-
lylic carbocation intermediate in Step I of the SQS reac-
tion. Step II of the SQS reaction requires that its highly re-
active carbocation intermediates be shielded from contact
with the aqueous solvent to prevent it from quenching the
reaction.This suggests that for Step II, the presqualene py-
rophosphate product of Step I moves deeper into the chan-
nel into a pocket that is lined with hydrophobic groups, in-
cluding Phe 288, whose mutation inactivates the enzyme.
This further suggests that Phe 288 functions to stabilize one
of the cationic intermediates in Step II (Fig. 25-54) through
–cation interactions.
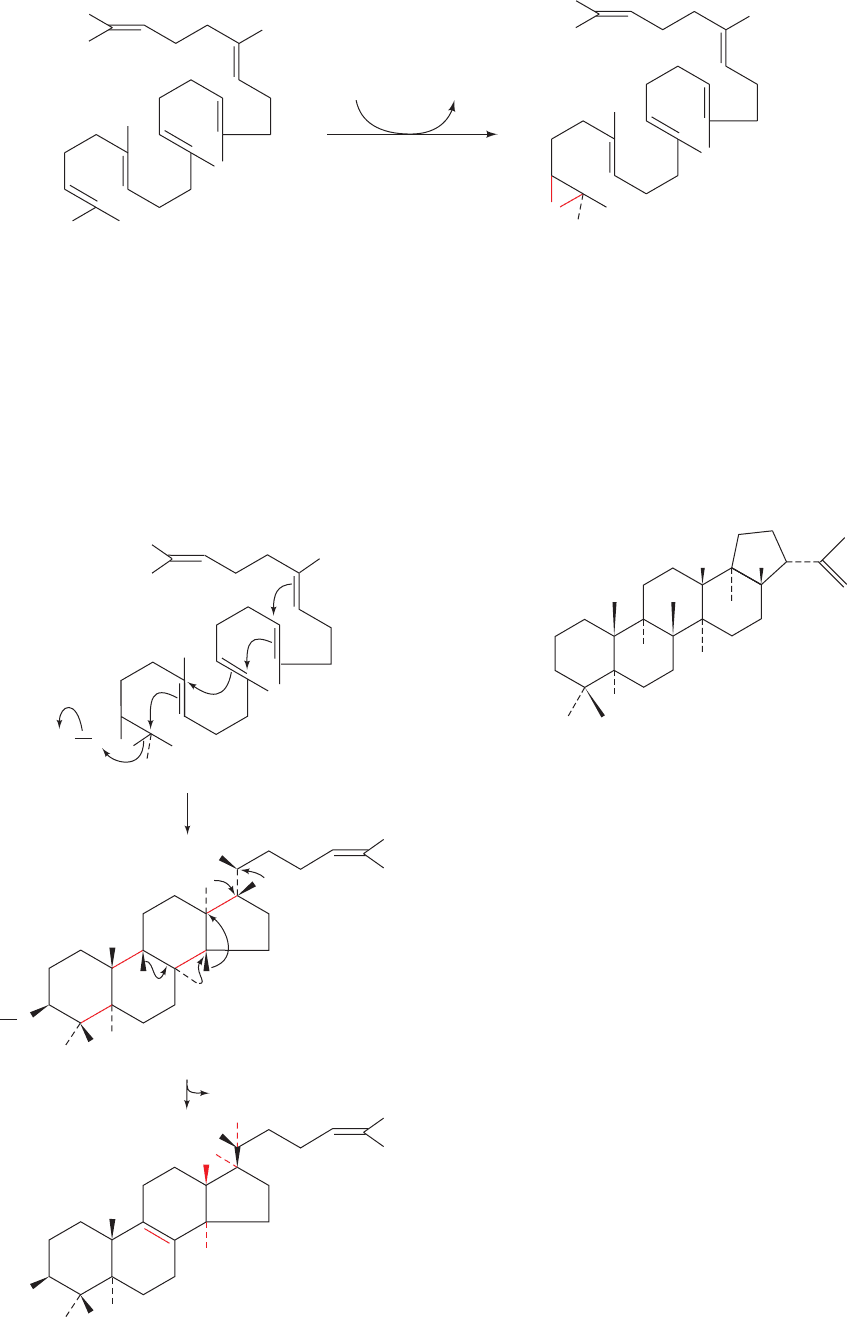
d. Lanosterol Is Produced by Squalene Cyclization
Squalene, an open-chain C
30
hydrocarbon, is cyclized to
form the tetracyclic steroid skeleton in two steps. Squalene
CP-320473
CH
2
CH
2
COO
COO
CH
2
C
O
CHNH
N
O
O
Cl
C(CH
3
)
3
epoxidase catalyzes the oxidation of squalene to form 2,3-
oxidosqualene (Fig. 25-56). Oxidosqualene cyclase (alter-
natively, lanosterol synthase) converts this epoxide to
lanosterol, the sterol precursor of cholesterol. The highly
exergonic reaction is a complex process involving cycliza-
tion of 2,3-oxidosqualene to a protosterol cation, via a
Class II mechanism involving protonation of the epoxide,
and rearrangement of this cation to lanosterol by a series
of 1,2 hydride and methyl shifts (Fig. 25-57). Note that this
reaction generates six of lanosterol’s seven chiral centers.
The X-ray structure of human oxidosqualene cyclase in
complex with lanosterol, determined by Armin Ruf, re-
veals that this 732-residue monomeric and monotopic [in-
tegral but not transmembrane] protein contains two struc-
turally similar domains named / barrels (Fig. 25-58a).
An / barrel consists of two concentric barrels of 6 helices
each, with the helices of the inner barrel largely parallel to
each other and antiparallel to those of the outer barrel
(much like an / barrel with the strands of the inner
barrel replaced by helices but with only 6 / units rather
than 8 / units). The enzyme’s active site is located inside
an elongated central cavity (Fig. 25-58b) to which the
lanosterol is bound and which is accessible from the mem-
brane via a nonpolar channel through the enzyme’s
membrane-immersed part.
The interactions of 2,3-oxidosqualene with oxidosqua-
lene cyclase cause it to fold and react such that it only
forms lanosterol.The active site cavity is lined with several
conserved aromatic side chains that are suitably positioned
to stabilize the catalyzed reaction’s intermediate carboca-
tions, while shielding the cationic intermediates from pre-
mature quenching by either enzyme nucleophiles or water.
The cationic cascade (Fig. 25-57) is initiated by proton do-
nation by Asp 455 to the epoxide ring of the prefolded 2,3-
oxidosqualene, and is quenched by His 232 acting as a gen-
eral base.
The importance of the proper placement of residues for
the formation of the correct product was demonstrated by
site-directed mutagenesis: The conversion of Thr 384 in
982 Chapter 25. Lipid Metabolism
Figure 25-55 X-ray structure of human squalene synthase
(SQS) in complex with the inhibitor CP-320473. This inhibitor
together with the side chains of D80, D84, Y171, and F288 are
drawn in ball-and-stick form colored according to atom type
(inhibitor C green, protein C cyan, N blue, O red, and Cl magenta).
The protein is viewed looking into its central channel with the
putative active sites for Steps I and II of the catalyzed reaction
at the bottom and top of the cleft, respectively. The protein’s
C-terminal transmembrane segment (residues 371–417) together
with its N-terminal 30 residues were excised to facilitate its
crystallization, which did not affect its in vitro catalytic activity.
[Based on an X-ray structure by Jayvardhan Pandit, Pfizer
Central Research, Groton, Connecticut. PDBid 1EZF.]
JWCL281_c25_940-1018.qxd 6/8/10 8:59 AM Page 982
oxidosqualene cyclase to Tyr causes some misplacement of
the C8“C9 double bond, with 11% of the product having
a C9“C11 double bond and 10% having a 9-hydroxy
group; the double mutation, T384Y/V454I, increases the
yield of the C9“C11 double bond to 64%. This, together
with different ways of substrate folding, explains how en-
zymes homologous to oxidosqualene cyclase cause oxi-
dosqualene and squalene to form different products. In-
deed, the bacterial enzyme squalene–hopene synthase, a
homolog of oxidosqualene cyclase, synthesizes the C
30
compound hopene
from squalene. Its X-ray structure in complex with 2-
azasqualene (squalene with its C2 atom replaced N), deter-
mined by Georg Schulz, reveals that the enzyme, which
structurally resembles oxidosqualene cyclase, folds squa-
lene in the configuration that is predicted to yield hopene.
In this case, the cationic cyclase cascade is initiated by the
protonation of a double bond instead of an epoxide.
Hopene
H
H
H
H
Section 25-6. Cholesterol Metabolism 983
Squalene
+ O
2
2,3-Oxidosqualene
+ H
2
O
squalene
epoxidase
NADPH NADP
+
O
O
Enz
H
H
H
H
H
2
2,3-Oxidosqualene
9
11
8
O
H
+
H
H
H
O
H
Protosterol cation
1
H
+
Lanosterol
Figure 25-56 The squalene epoxidase reaction.
Figure 25-57 The oxidosqualene cyclase reaction. (1) 2,3-
Oxidosqualene is cyclized to the protosterol cation in a process
that is initiated by the enzyme-mediated protonation of the
squalene epoxide oxygen while this extended molecule is folded
in the manner predicted by Bloch and Woodward. The opening
of the epoxide leaves an electron-deficient center whose
migration drives the series of cyclizations that ultimately form
the protosterol cation. (2) A series of methyl and hydride
migrations yields a hypothesized intermediate carbocation at C8,
which then eliminates a proton from C9 to form the C8“C9
double bond of lanosterol.
JWCL281_c25_940-1018.qxd 4/20/10 1:59 PM Page 983
e. Cholesterol Is Synthesized from Lanosterol
Conversion of lanosterol to cholesterol (Fig. 25-59) is a
19-step process that we shall not explore in detail. It in-
volves an oxidation and loss of three methyl groups. The
first methyl group is removed as formate and the other two
are eliminated as CO
2
in reactions that all require NADPH
and O
2
. The enzymes involved in this process are embed-
ded in the ER membrane.
f. Cholesterol Is Transported in the Blood and Taken
Up by Cells in Lipoprotein Complexes
Transport and cellular uptake of cholesterol are de-
scribed in Section 12-5.To recapitulate, cholesterol synthe-
sized by the liver is either converted to bile salts for use in
the digestive process (Section 25-1) or esterified by acyl-
CoA:cholesterol acyltransferase (ACAT) to form choles-
teryl esters
which are secreted into the bloodstream as part of the
lipoprotein complexes called very low density lipoproteins
(VLDL). As the VLDL circulate, their component triacyl-
Cholesteryl ester
H
H
H
C
O
R
O
984 Chapter 25. Lipid Metabolism
Figure 25-58 X-ray structure of human oxidosqualene cyclase
in complex with its product lanosterol. The monomeric protein is
shown in ribbon form (a) viewed along the axis of its / barrels,
and (b) rotated 90° about the horizontal axis with respect to Part
a. The inner helices of its two / barrels are cyan, its outer
helices are purple, its nonpolar membrane-immersed portion is
orange, and the remainder of the protein is pink.The lanosterol,
which occupies the enzyme’s centrally located active site cavity, is
shown in Part a in space-filling form and in Part b in stick form
embedded in its semitransparent space-filling form, both with C
green and O red.The catalytically important side chains of His
232 and Asp 455, together with molecules of the detergent
-octylglucoside and fragments of lipids that coat the enzyme’s
membrane-immersed portion, are drawn in stick form with side
chain C magenta, lipid and detergent C yellow, and O red. The
arrow in Part b points to the membrane-immersed opening of
the enzyme’s active site cavity. [Based on an X-ray structure by
Armin Ruf, F. Hoffmann-La Roche AG, Basel, Switzerland.
PDBid 1W6K.]
(a)
(b)
JWCL281_c25_940-1018.qxd 6/8/10 9:00 AM Page 984
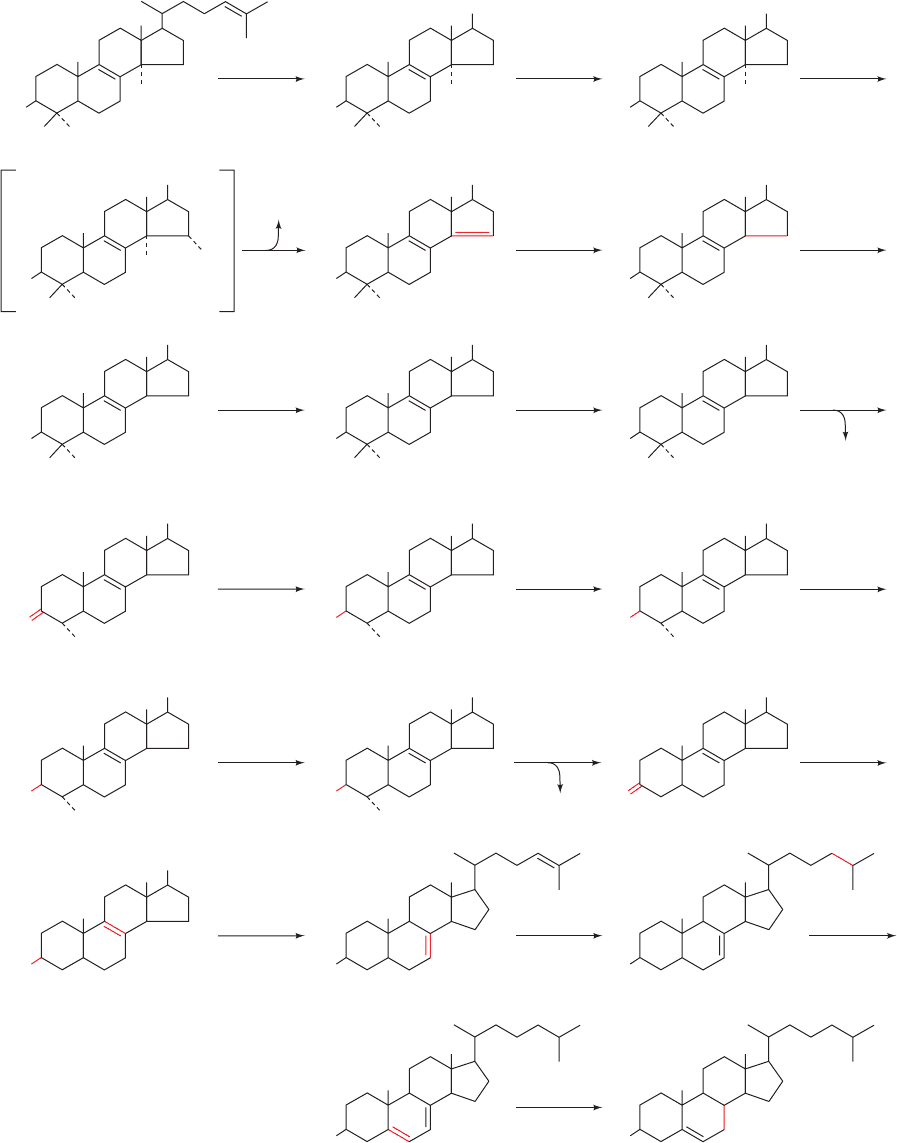
Section 25-6. Cholesterol Metabolism 985
Figure 25-59 The 19-reaction conversion of lanosterol to
cholesterol. [After Rilling, H.C. and Chayet, L.T., in Danielsson,
H. and Sjövall, J. (Eds.), Sterols and Bile Acids, p. 33, Elsevier
O
2
HO
1
Lanosterol
O
2
HO
CH
2
OH
RR
2
NADPH
NADPH
O
2
NAD(P)H
O
2
HO
CHO
3
NAD(P)H
HO
RR
5
NADPH
HO
R
CHO
OH
4
O
2
HO
6
NAD(P)H
O
2
HO
OCH HOOC
RR
8
NAD(P)H
CO
2
HO
9
NAD
+
HOH
2
C
R
HO
7
CO
2
NAD
+
NAD(P)H
O
2
HO
CH
2
OH
RR
11
NAD(P)H
O
2
NADPH
O
2
COOHCHO
HO
12
NAD(P)H
R
O
10
O
2
NAD(P)H
HO
RR
14
O
15
NADPH
R
HO
13
HCOOH
HO
17
R
HO
16
NADPH
HO
18
NADPH
HO
19
HO
20
Cholesterol
(1985), as modified by Bae, S.-H. and Paik,Y.-K., Biochem. J. 326,
609–616 (1997).]
JWCL281_c25_940-1018.qxd 4/20/10 1:59 PM Page 985
glycerols and most types of their apolipoproteins (Table
12-6) are removed in the capillaries of muscle and adipose
tissues, sequentially converting the VLDL to intermediate-
density lipoproteins (IDL) and then to low-density lipopro-
teins (LDL). Peripheral tissues normally obtain most
of their exogenous cholesterol from LDL by receptor-
mediated endocytosis (Fig. 25-60; Section 12-5Bc). Inside
the cell, cholesteryl esters are hydrolyzed by a lysosomal
lipase to free cholesterol, which is either incorporated into
cell membranes or reesterified by ACAT for storage as
cholesteryl ester droplets.
Dietary cholesterol, cholesteryl esters, and triacylglyc-
erols are transported in the blood by intestinally synthe-
sized lipoprotein complexes called chylomicrons. After re-
moval of their triacylglycerols at the peripheral tissues, the
resulting chylomicron remnants bind to specific liver cell
remnant receptors and are taken up by receptor-mediated
endocytosis in a manner similar to that of LDL.In the liver,
dietary cholesterol is either used in bile salt biosynthesis
(Section 25-6C) or packaged into VLDL for export. Liver
and peripheral tissues therefore have two ways of obtaining
cholesterol: They may either synthesize it from acetyl-CoA
986 Chapter 25. Lipid Metabolism
Endosome
Lysosome
Coated vesicle
Coated pit
LDL
particle
ApoB
protein
Golgi complex
LDL receptor
synthesis
Endoplasmic
reticulum
Nucleus
Cholesteryl
ester droplet
Amino acids
Recycling of
LDL receptors
LDL
receptors
Cholesteryl
ester
HMG-CoA reductase
synthesis
LDL receptor synthesis
ACAT
synthesis
Cholesterol
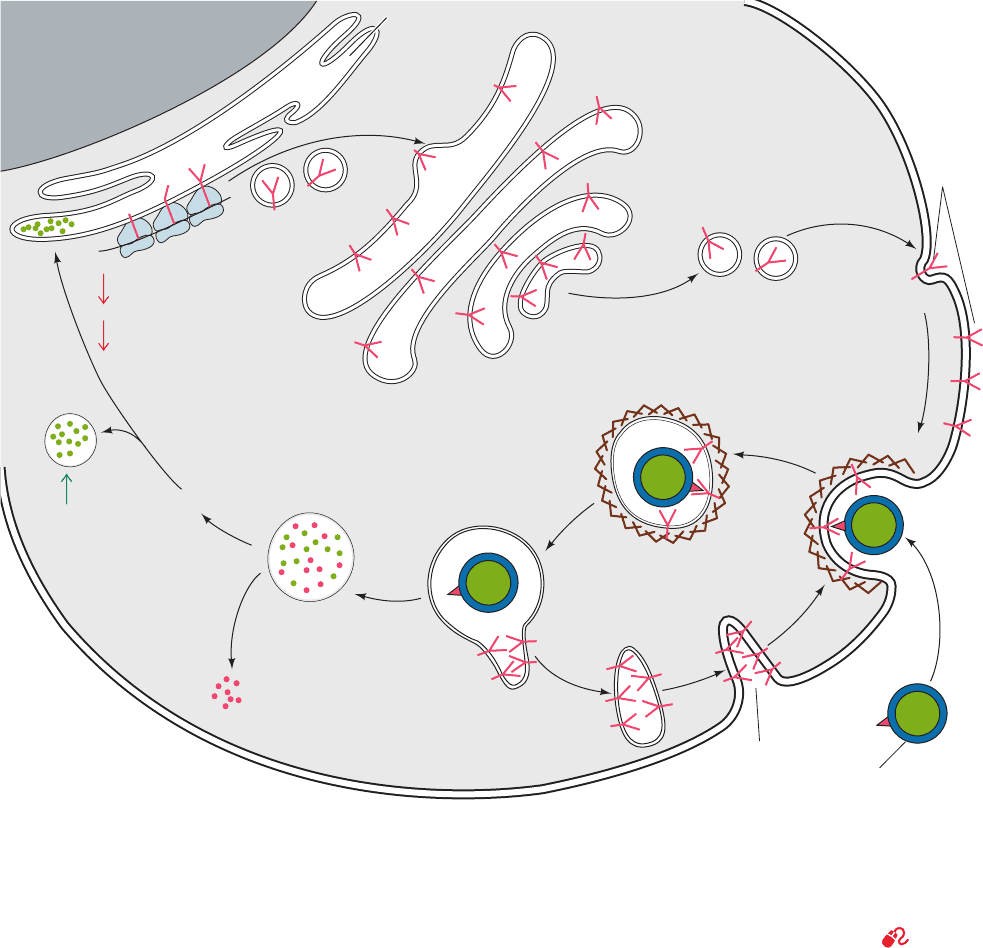
Figure 25-60 LDL receptor-mediated endocytosis in
mammalian cells. LDL receptor is synthesized on the
endoplasmic reticulum, processed in the Golgi complex, and
inserted into the plasma membrane as a component of coated
pits. LDL is specifically bound by the LDL receptor on the
coated pit and brought into the cell in endosomes that deliver
LDL to lysosomes while recycling LDL receptor to the plasma
membrane (Section 12-5Bc). Lysosomal degradation of LDL
releases cholesterol, whose presence decreases the rate of
synthesis of HMG-CoA reductase and LDL receptors (down
arrows) while increasing that of acyl-CoA:cholesterol
acyltransferase (ACAT; up arrow). [After Brown, M.S. and
Goldstein, J.L., Curr. Top. Cell. Reg. 26, 7 (1985).]
See the
Animated Figures
JWCL281_c25_940-1018.qxd 6/8/10 9:00 AM Page 986
by the de novo pathway we have just discussed, or they may
obtain it from the bloodstream by receptor-mediated endo-
cytosis. A small amount of cholesterol also enters cells by a
non-receptor-mediated pathway. Note however, that the
brain, which comprises ⬃2% of the human body mass but
contains ⬃30% of its cholesterol, must synthesize all of its
cholesterol because cholesterol cannot pass the blood-
brain barrier.
Cholesterol actually circulates back and forth between the
liver and peripheral tissues.While LDL transports cholesterol
from the liver, cholesterol is transported back to the liver by
high-density lipoproteins (HDL). Surplus cholesterol is dis-
posed of by the liver as bile salts, thereby protecting the body
from an overaccumulation of this water-insoluble substance.
B. Control of Cholesterol Biosynthesis
and Transport
Cholesterol biosynthesis and transport must be tightly reg-
ulated. There are three ways in which the cellular choles-
terol supply is maintained:
1. By regulating the activity of HMG-CoA reductase, the
enzyme catalyzing the rate-limiting step in the de novo choles-
terol biosynthesis pathway.This is accomplished in two ways:
(i) Short-term regulation of the enzyme’s catalytic ac-
tivity by (a) competitive inhibition, (b) allosteric ef-
fects, and (c) covalent modification involving re-
versible phosphorylation.
(ii) Long-term regulation of the enzyme’s concentra-
tion by modulating its rates of synthesis and
degradation.
2. By regulating the rate of LDL receptor synthesis, and
therefore the rate of cholesterol uptake. High intracellular
concentrations of cholesterol suppress LDL receptor syn-
thesis, whereas low cholesterol concentrations stimulate it.
3. By regulating the rate of esterification and hence the
removal of free cholesterol. ACAT, the enzyme that cat-
alyzes intracellular cholesterol esterification, is regulated
by reversible phosphorylation and by long-term control.
a. HMG-CoA Reductase Is the Primary Control
Site for Cholesterol Biosynthesis
HMG-CoA reductase is the rate-limiting enzyme in
cholesterol biosynthesis and, as therefore might be ex-
pected, constitutes the pathway’s main regulatory site. The
pathway branches after this reaction, however (Fig. 25-46);
ubiquinone, dolichol, farnesylated and geranylgeranylated
proteins, and isopentenyl adenosine are also essential,
albeit minor, products. HMG-CoA is therefore subject to
“multivalent” control, both long-term and short-term, in
order to coordinate the synthesis of all of these products.
b. Long-Term Feedback Regulation of HMG-CoA
Reductase Is Its Primary Means of Control
The main way in which HMG-CoA reductase is con-
trolled is by long-term feedback control of the amount of en-
zyme present in the cell. When either LDL–cholesterol or
mevalonate levels fall, the amount of HMG-CoA reduc-
tase present in the cell can rise as much as 200-fold, due to
an increase in enzyme synthesis combined with a decrease
in its degradation.When LDL–cholesterol or mevalonolac-
tone (an internal ester of mevalonate that is hydrolyzed to
mevalonate and metabolized in the cell)
are added back to a cell, these effects are reversed.
The mechanism by which cholesterol serves to control
the expression of the ⬎20 genes involved in its biosyn-
thesis and uptake, such as those encoding HMG-CoA re-
ductase and the LDL receptor, has been elucidated by
Michael Brown and Joseph Goldstein. These genes all
contain a DNA sequence upstream from the transcrip-
tion initiation site called the sterol regulatory element
(SRE). In order for these genes to be transcribed, a spe-
cific transcription factor, the sterol regulatory element
binding protein (SREBP), must bind to the SRE (eu-
karyotic gene expression is discussed in Section 34-3).
SREBP is synthesized as an integral membrane protein
that, when the cholesterol concentration is sufficiently
high (Fig. 25-61a), resides in the ER membrane in com-
plex with SREBP cleavage-activating protein (SCAP)
and a protein named Insig. SREBP (⬃1160 residues)
consists of three domains (Fig. 25-61a): (1) an ⬃480-
residue cytosolic N-terminal domain that is a member of
the basic helix–loop–helix/leucine zipper (bHLH/Z)
family of transcription factors (Section 34-3Br), which
specifically binds to SREs; (2) an ⬃90-residue transmem-
brane (TM) domain consisting of two TM helices con-
nected by an ⬃30-residue hydrophilic luminal loop; and
(3) an ⬃590-residue cytosolic C-terminal regulatory do-
main. SCAP (1276 residues) consists of two domains: (1)
a 730-residue N-terminal domain that contains eight TM
helices; and (2) a 546-residue cytosolic C-terminal do-
main that contains five copies of the protein–protein in-
teraction motif known as a WD repeat (also called a
WD40 sequence motif because it is ⬃40 residues long;
Section 19-2Cc) and which presumably forms a 5-bladed
 propeller similar to the 7-bladed  propeller of the
G

subunit (Fig. 19-19b). Insig (named to denote insulin-
induced gene, although the insulin effect was later found
to be indirect) consists mainly of six TM helices. SCAP
and SREBP associate through the interaction of the
SCAP’s regulatory domain with SREBP’s WD domain
(Fig. 25-61a).
SCAP functions as a sterol sensor. An ⬃170-residue
segment of its TM domain, the sterol-sensing domain, in-
teracts with sterols although how it does so is unknown.
When the cholesterol in the ER membrane is depleted
(Fig. 25-61b), SCAP changes conformation and then
Mevalonolactone
C
OC
O
CH
2
HO
CH
2
CH
2
H
3
C
Section 25-6. Cholesterol Metabolism 987
JWCL281_c25_940-1018.qxd 6/18/10 7:51 AM Page 987
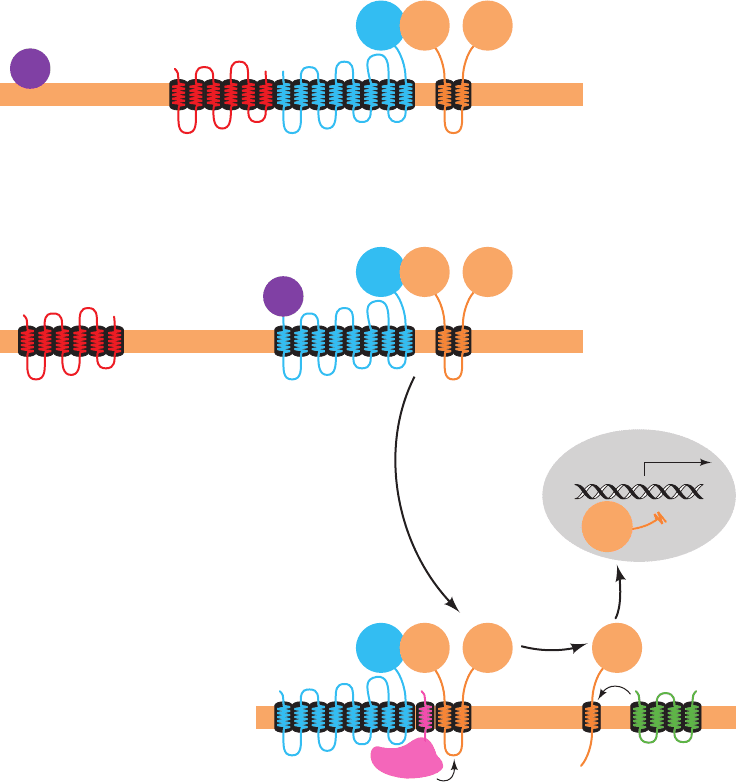
releases Insig, which allows SCAP to bind to the Sec24
cargo-binding subunit of the COPII coated vesicle protein
(Section 12-4Ca).The COPII vesicle then escorts its bound
SREBP–SCAP complex to the Golgi apparatus.
In the Golgi apparatus, SREBP is sequentially cleaved
by two membrane-bound proteases (Fig. 25-61b). Site-1
protease (S1P), a serine protease of the subtilisin family,
cleaves SREBP in the luminal loop that connects its two
transmembrane helices but only when it is associated
with SCAP.This cleavage exposes a peptide bond located
3 residues from the beginning of SREBP’s N-terminal
TM helix to cleavage by site-2 protease (S2P), a zinc met-
alloprotease. This releases the bHLH/Z domain to mi-
grate to the nucleus where it activates the transcription
of its target genes. The cholesterol level in the cell
thereby rises until SCAP no longer induces the transloca-
tion of SREBP to the Golgi, a classic case of feedback in-
hibition.
This complex regulatory pathway was elucidated, in
part, through the generation of several lines of transgenic
mice that overexpress one or another of the foregoing pro-
teins, and knockout mice that lack one or another of these
988 Chapter 25. Lipid Metabolism
SREBPSCAP
Cytosol
ER
COPII
Insig
Transport
to Golgi
Transport
to nucleus
Golgi
apparatus
Lumen
Nucleus
SRE
WD
S1P
S2P
Serine protease Metalloprotease
Zn
2+
bHLH
bHLHReg.
SREBPSCAP
Cytosol
ER
Insig
Lumen
COPII
High cholesterol
Low cholesterol
WD bHLHReg.
WD bHLHReg. bHLH
(a)
(b)
Figure 25-61 The cholesterol-mediated proteolytic activation
of SREBP. (a) When cholesterol levels in the cell are high, the
Insig–SCAP–SREBP complex resides in the ER membrane.
(b) When cholesterol levels are low, SCAP releases Insig and binds
COPII protein.The SCAP–SREBP complex is then transported,
via COPII-coated vesicles, to the Golgi apparatus, where SREBP
undergoes sequential proteolytic cleavage by the
membrane-bound proteases S1P and S2P. This releases SREBP’s
bHLH/Z-containing N-terminal domain, which enters the
nucleus where it binds to the SREs of its target genes, thereby
inducing their transcription. [After Goldstein, J., Rawson, R.B.,
and Brown, M., Arch. Biochem. Biophys. 397, 139 (2002).]
JWCL281_c25_940-1018.qxd 8/9/10 10:21 AM Page 988
proteins. For example, knockout mice lacking either SCAP
or S1P in their livers have decreased expression of both
HMG-CoA reductase and LDL receptor, even when fed a
cholesterol-deficient diet. In contrast, transgenic mice that
overexpress SREBP or SCAP have greatly increased ex-
pression of the foregoing proteins. In fact, animals overpro-
ducing only the bHLH/Z domain of SREBP have mas-
sively enlarged livers (up to 4-fold larger than normal) due
to engorgement with triacylglycerols and cholesteryl esters
and yet they continue to transcribe SREBP’s target genes
such that their mRNA levels are up to 75-fold greater than
normal. Many individuals suffering from obesity or dia-
betes caused by insulin resistance (type 2 diabetes; Section
27-4B) have fatty livers, which in some cases leads to liver
failure. Fatty livers due to insulin resistance appear to be
caused by elevated levels of SREBP in response to ele-
vated insulin levels.
The level of HMG-CoA reductase also responds to
the level of the cholesterol precursor lanosterol (Section
25-6Ad). HMG-CoA reductase’s ER membrane-bound
N-terminal domain contains eight TM helices, whereas its
C-terminal domain, which contains its active site and is
linked to the N-terminal domain via a flexible Pro-rich se-
quence, projects into the cytosol. Insig binds to an enzy-
matic complex that marks proteins for degradation by co-
valently linking them to the protein ubiquitin (Section
32-6B). When lanosterol accumulates in the ER mem-
brane, the N-terminal domain of HMG-CoA reductase
also binds to Insig,and is thus marked for destruction. Con-
sequently, the 12-hour half-life of HMG-CoA reductase
in sterol-deprived cells decreases to 1 hour when sterols
are plentiful.
c. Regulation of HMG-CoA Reductase by Covalent
Modification Is a Means of Cellular Energy
Conservation
HMG-CoA reductase exists in interconvertible more ac-
tive and less active forms, as do glycogen phosphorylase
(Section 18-3Ca), glycogen synthase (Section 18-3D), pyru-
vate dehydrogenase (Section 21-2Cb), and acetyl-CoA car-
boxylase (Section 25-4Ba), among others. The unmodified
form of HMG-CoA reductase is more active and the phos-
phorylated form is less active. HMG-CoA reductase is
phosphorylated (inactivated) at its Ser 871 in a bicyclic cas-
cade system by the covalently modifiable enzyme AMP-
dependent protein kinase (AMPK), which, as we saw in
Section 25-4Ba, also acts on acetyl-CoA carboxylase [in
this context, this enzyme was originally named HMG-CoA
reductase kinase (RK), until it was found to be identical to
AMPK]. It appears that this control is exerted to conserve
energy when ATP levels fall and AMP levels rise, by in-
hibiting biosynthetic pathways. This hypothesis was tested
by Brown and Goldstein, who used genetic engineering
techniques to produce hamster cells containing a mutant
HMG-CoA reductase with Ala replacing Ser 871 and
therefore incapable of phosphorylation control.These cells
respond normally to feedback regulation of cholesterol
biosynthesis by LDL–cholesterol and mevalonate but, un-
Section 25-6. Cholesterol Metabolism 989
like normal cells, do not decrease their synthesis of choles-
terol on ATP depletion, supporting the idea that control of
HMG-CoA reductase by phosphorylation is involved in
energy conservation.
d. LDL Receptor Activity Controls
Cholesterol Homeostasis
LDL receptors clearly play an important role in the
maintenance of plasma LDL–cholesterol levels. In normal
individuals, about half of the IDL formed from the VLDL
reenters the liver through LDL receptor-mediated endocy-
tosis (IDL and LDL both contain apolipoproteins that
specifically bind to the LDL receptor; Section 12-5Bc).The
remaining IDL are converted to LDL (Fig. 25-62a). The
serum concentration of LDL therefore depends on the rate
at which liver removes IDL from the circulation, which, in
turn, depends on the number of functioning LDL receptors
on the liver cell surface.
High blood cholesterol (hypercholesterolemia), which
results from the overproduction and/or underutilization of
LDL, is known to be caused by either of two metabolic
irregularities: (1) the genetic disease familial hyper-
cholesterolemia (FH) or (2) the consumption of a high-
cholesterol diet. FH is a dominant genetic defect that
results in a deficiency of functional LDL receptors (Section
12-5Ca). Homozygotes for this disorder lack functional
LDL receptors, so their cells can absorb neither IDL nor
LDL by receptor-mediated endocytosis.The increased con-
centration of IDL in the bloodstream leads to a correspon-
ding increase in LDL, which is, of course, underutilized
since it cannot be taken up by the cells (Fig. 25-62b). FH
homozygotes therefore have plasma LDL–cholesterol lev-
els three to five times higher than average. FH heterozy-
gotes, which are far more common, have about half of the
normal number of functional LDL receptors and plasma
LDL–cholesterol levels about twice the average.
The long-term ingestion of a high-cholesterol diet has an ef-
fect similar, although not as extreme, as FH (Fig. 25-62c). Ex-
cessive dietary cholesterol enters the liver cells in chylomi-
cron remnants and represses the synthesis of LDL receptor
protein. The resulting insufficiency of LDL receptors on the
liver cell surface has consequences similar to those of FH.
LDL receptor deficiency, whether of genetic or dietary
origin, raises the LDL level by two mechanisms: (1) in-
creased LDL production resulting from decreased IDL
uptake and (2) decreased LDL uptake. Two strategies for
reversing these conditions (besides maintaining a low-
cholesterol diet) are being used in humans:
1. Ingestion of anion exchange resins (Section 6-3A) that
bind bile salts, thereby preventing their intestinal absorption
(resins are insoluble in water). Bile salts, which are derived
from cholesterol, are normally efficiently recycled by the
liver (Section 25-6C). Elimination of resin-bound bile salts in
the feces forces the liver to convert more cholesterol to bile
salts than otherwise. The consequent decrease in the serum
cholesterol concentration induces synthesis of LDL recep-
tors (of course, not in FH homozygotes). Unfortunately, the
JWCL281_c25_940-1018.qxd 4/20/10 1:59 PM Page 989
decreased serum cholesterol level also induces the synthesis
of HMG-CoA reductase, which increases the rate of choles-
terol biosynthesis. Ingestion of bile salt–binding resins such
as cholestyramine (sold as Questran) therefore provides only
a 15 to 20% drop in serum cholesterol levels.
2. Treatment with competitive inhibitors of HMG-CoA
reductase. These include (Fig. 25-63) the fungal derivatives
lovastatin (also called mevinolin and sold as Mevacor),
pravastatin (Pravachol), and simvastatin (Zocor) as well as
the synthetic inhibitor atorvastatin (Lipitor), compounds
990 Chapter 25. Lipid Metabolism
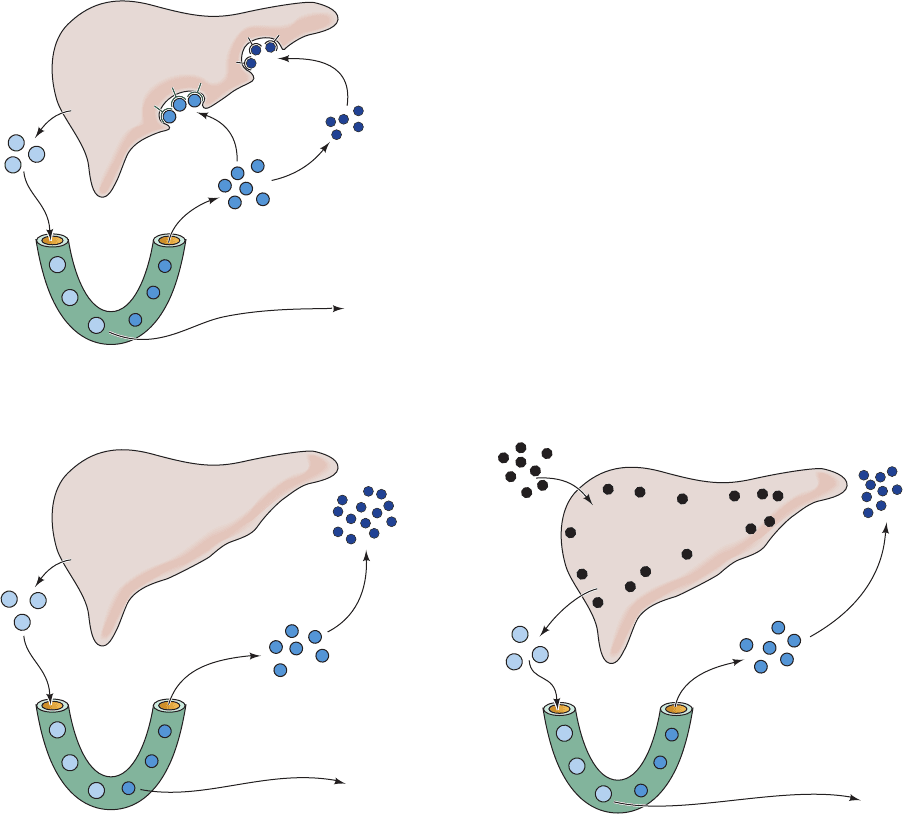
Figure 25-62 Control of plasma LDL production and uptake
by liver LDL receptors. (a) In normal human subjects, VLDL is
secreted by the liver and converted to IDL in the capillaries of
the peripheral tissues.About half of the plasma IDL particles
bind to the LDL receptor and are taken up by the liver.The
remainder are converted to LDL at the peripheral tissues. (b) In
individuals with familial hypercholesterolemia (FH), liver LDL
receptors are diminished or eliminated because of a genetic
Liver
VLDL
LDL
IDL
VLDL
Dietary
cholesterol
LDL
IDL
Capillary
lipoprotein
lipase
Free fatty
acids
(a) Normal
(c) High cholesterol diet
Normal LDL
receptor
function
Cholesterol repression
of LDL receptor
synthesis
VLDL
LDL
IDL
(b) Familial hypercholesterolemia
Genetically defective
LDL receptor
synthesis
lipoprotein
lipase
Free fatty
acids
lipoprotein
lipase
Free fatty
acids
defect. (c) In normal individuals who have a long-term
high-cholesterol diet, the liver is filled with cholesterol, which
represses the rate of LDL receptor production. Receptor
deficiency, whether of genetic or dietary cause, raises the plasma
LDL level by increasing the rate of LDL production and
decreasing the rate of LDL uptake. [After Goldstein, J.L. and
Brown, M.S., J. Lipid Res. 25, 1457 (1984).]
JWCL281_c25_940-1018.qxd 4/20/10 1:59 PM Page 990