Schlick T. Molecular Modeling and Simulation: An Interdisciplinary Guide
Подождите немного. Документ загружается.

140 5. Nucleic Acids Structure Minitutorial
5.2.5 Stabilizing Polynucleotide Interactions
Both hydrogen bonding and base stacking are considered to be intrinsic factors
for helix stability. The estimates given for intrinsic energies of hydrogen-bonding
and base-pair stacking in Chapter 7 (Subsection 7.2.1) suggest that the latter is at
least as important for overall helix stability.
The bases in DNA double helices are normally linked by hydrogen bonds
in the WC bps as depicted in Figure 5.3. Recall that in this weak noncovalent
electrostatic interaction a hydrogen atom is shared between negatively-charged
donor and acceptor atoms, typically nitrogens or oxygens in biological polymers.
Besides WC, other hydrogen-bonding patterns involve variations in the position-
ing of the two bases relative to one another and in the hydrogen-bond defining
atoms.
Base-pair stacking refers to favorable interactions between hydrogen-bonded
neighboring bps (imagine weak interactions between the stacked ladder rungs).
These arise from favorable van der Waals and hydrophobic contacts, which can
optimize the water-insoluble areas of contact. Another factor in stability is electro-
statics, which leads to preferred distributions of the partial electric charges spread
over the aromatic rings. In addition, specific hydrogen bonds, other than classic
WC bps, can form between adjacent bps if the bps are properly oriented. One
example is propeller-twisting of base pairs (introduced below).
In fact, we are growing to appreciate the many hydrogen-bonding and base-
pair stacking arrangements that one or more polynucleotide strands can form,
including residues with modified bases, bases with bound adducts, and peptide
nucleic acid structures [737,907,1191,1216].
As will be further discussed, the notion of a perfectly ordered double-helical
molecules is thus just an ideal reference. Many of the biological functions of
DNA and DNA/protein complexes require appreciation of the sequence-specific,
atomic-level variations.
5.2.6 Chain Notation
A DNA chain has polarity. Each strand of the chain has a terminal C5
–OH group
on one end and a terminal C3
–OH group on the other. Thus, the two intertwined
strands run in anti-parallel directions. Conventionally, the base sequence in a
polynucleotide is specified for the 5
→ 3
direction; the complementary strand is
automatic for ideal WC bps.
To number the 2n bases and n base pairs in a DNA duplex, Strand I — the
sequence strand — and Strand II — its antiparallel companion — are specified,
as shown in Figure 5.1. Bases along Strand I are numbered 1 to n in the 5
→ 3
direction, and bases along Strand II are numbered from n +1to 2n,alsointhe
5
→ 3
direction. Base pairs 1 to n are numbered so that they coincide with the
bases of Strand I. Thus, bp 1 involves base 1 of Strand I with base 2n of Strand II;
bp 2 is the hydrogen-bonded pair of Strand I’s base 2 with Strand II’s base 2n−1;
and so on.
5.2. The Basic Building Blocks of Nucleic Acids 141
Nucleosides are abbreviated by a pair of letters: a lower-case Roman letter
denotes the sugar type (“r” or “d”), and an upper-case Roman letter repre-
sents the base type (C, G, A, T, U). For example, rA is adenosine — ribose
plus adenine, and dC is deoxycytidine — deoxyribose plus cytosine. The base
prefixes are often dropped when the type of sugar is obvious. Nucleotides
are abbreviated by adding a lower-case “p” (for phosphate) to the nucleoside
symbol.
Thus, the sequence dGpdCpdApdC is a tetramer in a DNA double helix where
GisattheC5
–OH end and C is at the C3
–OH end. For brevity, these lower-
case “p”s are often omitted when a specific nucleic-acid sequence is specified.
In the nucleic acid literature, duplex units of DNA for 2–12 base pairs are com-
monly termed as dimer, trimer, tetramer, pentamer, hexamer, heptamer, octamer,
nonamer, decamer, undecamer, and dodecamer, respectively.
5.2.7 Atomic Labeling
Standard atomic numbering schemes have been recommended for nucleic acids
[1080], as follows (see Figures 5.3 and 5.4):
• Sugar atoms are distinguished from base atoms by a prime suffix, and
within the sugar the numbering sequence is counted clockwise from the
ring oxygen in the direction of the carbon attached to the base nitrogen:
O4
→ C1
→ C2
→ C3
→ C4
→ O4
.
• In the polynucleotide backbone, the counting direction for torsion angles
(α, β, γ, δ, and ζ) is specified by the sequence:
P → O5
→ C5
→ C4
→ C3
→ O3
→ P.
• Base atoms are numbered systematically as shown in Figure 5.3. The pro-
cedure for labeling base atoms is different than that used for the sugar:
Whereas the sugar atoms are numbered by atom types (O4
,C1
through
C4
), the base atoms are numbered according to position in the ring (in
cytosine, for example, N1 and N3 are the two ring nitrogens, and C2, C4,
C5 and C6 are the four ring carbons).
One nitrogen of the base is always connected to the C1
of the sugar
by the glycosyl bond (N to C1
). Thus, according to the base numbering
scheme, a pyrimidine base attached the glycosyl nitrogen has N1–C1
,and
a purine base has N9–C1
.
• When more than one oxygen or hydrogen are attached to the same atom,
the two substituents are generally distinguished by the numbers 1 and 2.
This applies to the two C5
hydrogens, H1 and H2, and the two oxygens at
P, O1P and O2P. (Note that O2 is defined also for atoms of the pyrimidine
bases). The order of these substituents is such that when we look along the
counting direction of the main chain or the counting direction of the sugar
ring, a clockwise direction gives substituent 1 and then 2. For example,
142 5. Nucleic Acids Structure Minitutorial
when we look along the O5
→ C5
bond, a clockwise counting gives sub-
stituents C4
,H1,andH2ofatomC5
(the hydrogens are generally labeled
H5
1andH5
2).
Table 5.2. Nucleic acid torsion angle definitions.
Angle Sequence Angle Sequence
α O3
–P–O5
–C5
τ
0
C4
–O4
–C1
–C2
β P–O5
–C5
–C4
τ
1
O4
–C1
–C2
–C3
γ O5
–C5
–C4
–C3
τ
2
C1
–C2
–C3
–C4
δ C5
–C4
–C3
–O3
τ
3
C2
–C3
–C4
–O4
C4
–C3
–O3
–P τ
4
C3
–C4
–O4
–C1
ζ C3
–O3
–P–O5
χ O4
–C1
–N1–C2 (pyrimidine)
O4
–C1
–N9–C4 (purine)
5.2.8 Torsion Angle Labeling
Recall that a torsion angle describes the relative orientation of a bonded 4-atom
sequence i–j–k–l as the angle between the two planes defined by i–j–k and j–k–l
(see Box 3.4 and Figure 3.14 in Chapter 3). As with polypeptides, the torsion
angles in polynucleotide chains are denoted by a systematic set of Greek letters,
as shown in Figure 5.4 and defined in Table 5.2. For the backbone rotations, the
letters α, β, γ, δ, and ζ are used, in the order along the chain backbone direction.
For the sugar molecule, the endocyclic (i.e., ring) torsion angles are designated
by τ
0
through τ
4
along the ring direction noted above (clockwise) so that τ
j
is
the torsion angle for the sequence C
j
−1
–C
j
–C
j
+1
–C
j
+2
where C
0
is O4
and
the integer j
is equal to j modulo 5 (hence C
5
is O4
and C
−1
is C4
, for example;
see Table 5.2)[35].
For describing the orientation of the nitrogenous base to the sugar, the torsion
angle χ is defined about the glycosyl bond; χ is the torsion angle of the O4
–C1
–
N1–C2 sequence for a pyrimidine base and the torsion angle of O4
–C1
–N9–C4
for a purine base. Rotational sequences for the nitrogenous bases are not given
special symbols, because the bases are approximately planar.
5.3 Nucleic Acid Conformational Flexibility
As in polypeptides, where conformational flexibility is limited due to steric hin-
drance (as described, for example, in the Ramachandran plots introduced in
Chapter 3), the observed set of conformations in nucleic acids is limited by
energetic and chemical considerations. A description of these conformations is
more complex than those for the backbone φ and ψ pairs and secondary-structure
5.3. Nucleic Acid Conformational Flexibility 143
elements as used for polypeptides, since a greater conformational variability exists
in nucleic acid residues, with additional variation in helical parameters. Sequence
and environmentalfactors (e.g., cations, bound ligands) affect sensitively the local
structural fluctuations.
5.3.1 The Furanose Ring
The 5-membered furanose sugar ring is generally nonplanar in nucleic acids. One
or two atoms may pucker out of the plane defined by the remaining skeletal ring
atoms. When four ring atoms are planar, the pucker form is called envelope;when
two atoms pucker at opposite sides of the plane defined by the remaining three
ring atoms, the conformation is known as a twist (see Figure 5.5).
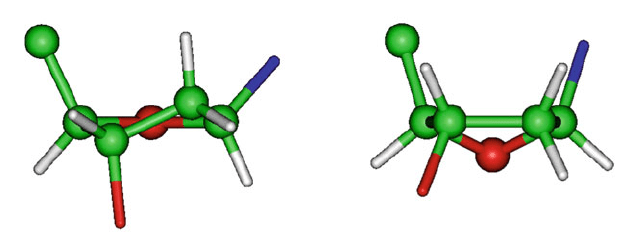
The sugar pucker type is described by the atom or atoms that deviate from
that three or four-atom ring plane. Atoms displaced on the same side of C5
are
designated as endo, and atoms displaced on the opposite side of C5
are called exo.
C2'
C3'
C5'
O4'
O3'
N
C1'C4'
C2'-endo / C3'-exo
C2'C3'
C5'
O3'
N
O4'
C1'C4'
O4'-exo
Figure 5.5. Envelope and twist puckering forms for a 5-membered ring: C2
–endo/C3
–exo
symmetric twist (left) and O4
-exo envelope (right); for clarity, hydrogens attached to
exocyclic atoms are not shown.
To describe nucleic acid sugar conformations, it is convenient to use a
pseudorotation path developed on the basis of the 5-membered carbon ring cy-
clopentane [35,271]. In cyclopentane, a wavelike motion between conformations
of equal energy can be imagined with respect to a mean plane. This mean ring
plane can be defined (in various ways) by positions of the five skeletal ring atoms.
In both the Altona/Sundaralingam (AS) [35] and Cremer/Pople (CP) [271]de-
scriptions, this sinusoidal motion is described by a wave amplitude and phase
shift: (q, φ) and (τ
max
,P), respectively, as follows.
In the AS description, the five endocyclic torsion angles are restricted to the
values:
τ
j
= τ
max
cos [ P +4π/5(j − 2) ] ,j=0, 1, 2, 3, 4. (5.1)
144 5. Nucleic Acids Structure Minitutorial
In CP, the five perpendicular displacements of the ring carbons from a mean plane
are described by the cosine series:
z
j
=(2/5)
1/2
q cos [ φ +4π/5(j − 2) ] ,j=0, 1, 2, 3, 4. (5.2)
The two formalisms are only equivalent under the assumption of infinitesimal
displacements from a regular planar pentagon. A more general relation based on
a simple analysis of model riboses was derived in [522].
The wave-like pseudorotation path described by eq. (5.1)defines10envelope
conformations for P =18
o
, 54
o
, 90
o
,...,342
o
and 10 twist conformations for
P =0
o
, 36
o
, 72
o
,...,324
o
. Figure 5.6 shows the 10 envelopes and two of the
10 twists. The quadrant terminology of N, S, E, and W is often used to describe
North, South, East, and West sugar-pucker regions.
P
0
18
54
90
126
162
180
198
234
270
306
342
c2
’
−exo
c1’−endo
c3’−end
o
c
4’
−exo
o4
’
−endo
o4
’
−exo
c
4’−en
d
o
c3’−e
x
o
c2’
−endo
c1’−exo
NORTH
W
E
S
T
SOUTH
E
A
S
T
Figure 5.6. Sugar pseudorotation cycle.
Using this description for nucleic-acid sugars, we see from Figure 5.5 that
O4
–exo, for example, is an envelope conformation with O4
puckering out of
the C4
–C3
–C2
–C1
plane on the opposite side of C5
. Similarly, C2
–endo/
C3
–exo is a twist conformation with C2
puckering out of the C4
–O4
–C1
plane
on the same side of C5
and C3
puckering out of the same plane on the opposite
side of C5
.
Two major types of puckering modes are observed in nucleic acid sugars.
They cluster around the North C3
–endo (0
o
<P<36
o
) and South C2
–endo
5.3. Nucleic Acid Conformational Flexibility 145
(144
o
<P <188
o
) puckers; see Figure 5.7. The puckering amplitude τ
max
averages around 40
o
± 5
o
. Still, significant deviations of sugar pseudorotation
parameters are commonly observed [1080], especially as new forms of DNA are
being discovered.
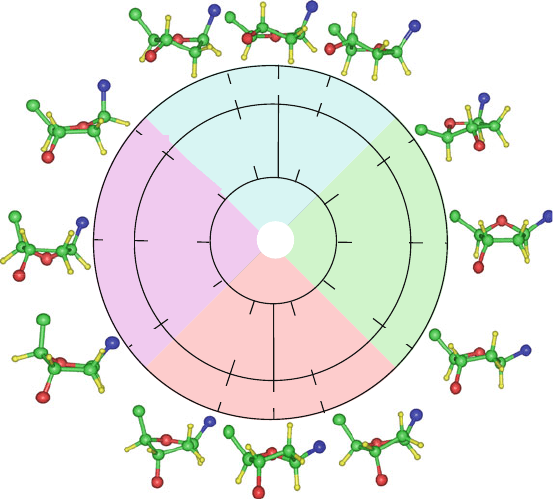
Figure 5.8 shows sugar puckering patterns of crystallographically deter-
mined nucleosides and nucleotides. Such analyses are possible using the above
(Fourier) formalism extrapolated from cyclopentane motions. The numerical val-
ues for the two Fourier parameters are estimated by the Cartesian coordinates of
atomic-resolution models.
C2'-endoC3'-endo
Anti
Syn
C2'
C2'
C2'
C2'
C3'
C3'
C3'
C3'
χ
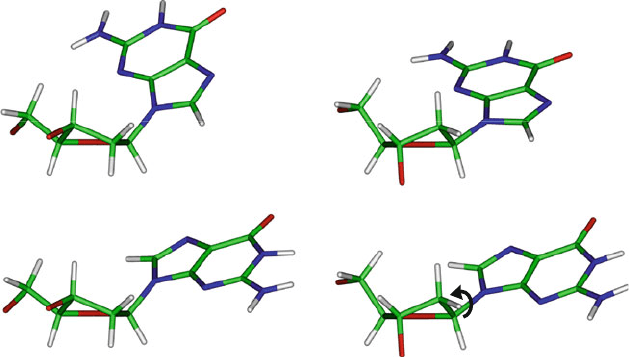
Figure 5.7. The common C3
–endo (left) and C2
–endo (right) sugar puckers for a
deoxyguanosine in combination with the glycosyl torsion angle in the syn (top) and anti
(bottom) conformations. Note that the 6-membered ring of G lies over the sugar in syn and
away in anti.
The study of nucleic acid sugar conformationsand conformational interchanges
has been an active area of research, beginning with the pioneering work of Levitt
and Warshel [750] (see works cited in [59]), since the sugar conformationstrongly
affects the overall helical structure.
5.3.2 Backbone Torsional Flexibility
In addition to the five internal sugar torsions described above, the six phospho-
diester backbone torsion angles α, β, γ, δ, ,andζ are flexible but restricted to
sterically allowable regions. Different values are also characteristic of various
helical structures, as shown in Table 5.4 (discussed later).
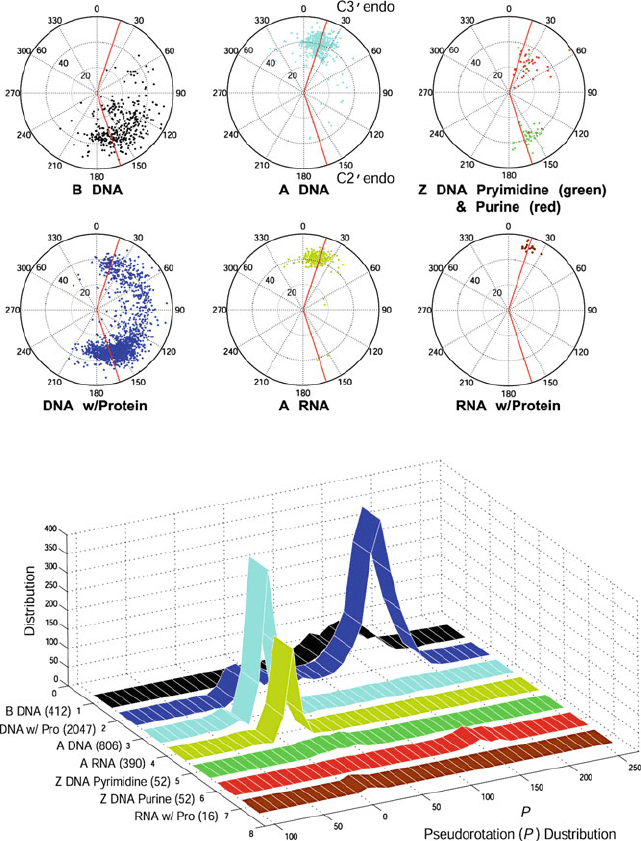
146 5. Nucleic Acids Structure Minitutorial
Figure 5.8. Sugar pucker conformations analyzed from the NDB for higher-resolution
structures (<2
˚
A) released as of July 31, 2009 and analyzed from the website of the
3DNA program (http://w3dna.rutgers.edu/).In the averaging, two nucleotides from each
end are excluded. The total number of residues analyzed to obtain sugar parameters for
A-DNA, B-DNA, Z-DNA pyrimidines, Z-DNA purines, A-RNA, DNA/Protein complexes,
and RNA/Protein complexes is indicated in the bottom graph (in parentheses). In the sugar
wheels, the radial coordinate is the phase amplitude τ
max
and the angle is the phase an-
gle of pseudorotation (P ). All the radial plots are colored monochromatically except for
Z-DNA, where two colors are used for pyrimidines and purines.
5.3. Nucleic Acid Conformational Flexibility 147
Recall that a torsion angle range can be described by the three exhaustive
regions of the conformational space — gauche
+
(g
+
), gauche
−
(g
−
), and trans (t)
(see also Figure 3.13 in Chapter 3). In terms of these classifications, both α (about
P–O5
)andγ (about C5
–C4
) exhibit relatively large flexibility, with the g
+
,g
−
,
and t positions observed. The angles β (about O5
–C5
)and (about C3
–O3
)
tend to cluster around the trans state, with occupying a broad t/g
−
range. The
backbone torsion angle δ (about C4
–C3
) is correlated to the sugar pseudorota-
tion puckering state, since the sugar torsion τ
3
is defined about the same C–C
bond. This correlation can be described roughly by δ = τ
3
+ 120
o
. Finally,
ζ (about O3
–P) is rather flexible, with all three ranges observed.
See Figure 5.9 for distributions of these backbone torsion angles, as analyzed
for the same high-resolution crystal structures used to generate Figure 5.8.
0
–90
180
90
α
β
γ
δ
ε
ζ
χ
B-DNA
0
–90
–90
180
90
α
β
γ
δ
ε
ζ
χ
DNA w/Protein
α
β
γ
δ
ε
ζ
χ
A-DNA
α
β
γ
δ
ε
ζ
χ
A-RNA
α
β
γ
δ
ε
ζ
χ
Z DNA Pyrimidine
Z DNA Purine
0
–90
180
90
0
180
90
0
–90
180
90
0
–90
180
90
0
–90
180
90
α
β
γ
δ
ε
ζ
χ
α
β
γ
δ
ε
ζ
χ
RNA w/ Protein
γ
α
Figure 5.9. Observed ranges of nucleic-acid torsion angles for higher-resolution structures
(<2
˚
A) for the same residues analyzed for Figure 5.8.

148 5. Nucleic Acids Structure Minitutorial
5.3.3 The Glycosyl Rotation
Relative to the sugar moiety, the base can assume two major orientations about
the glycosyl C1
–N bond: syn and anti (torsion angles of 0 and 180
o
, respec-
tively) [1080]. Roughly speaking, four major conformations are favored. They
correspond to the combinations of C3
–endo and C2
–endo sugar puckers with
syn and anti values for χ. Favored combinations of {P , χ} pairs vary for the dif-
ferent nucleosides or nucleotides. They depend on the chemical structure of the
sugar, the size of the base, and the nature of the nucleoside substituents (chemi-
cal derivatives). For example, deoxyribose nucleosides and nucleotides prefer the
C2
–endo conformation over C3
–endo [944]. In pyrimidine nucleotides, the anti
orientation of χ about the sugar ring is finely tuned by the sugar pucker [1303].
In Figure 5.7 the two orientations of syn and anti bases are illustrated
for deoxyguanosine in combination with the two common sugar puckers. The
{C3
–endo, syn} combination of this figure (top left) is that observed in the purine
of Z-DNA helices while the {C2
–endo, anti} combination (bottom right) is typ-
ically observed in the B-DNA varieties (and in Z-DNA pyrimidines). Figure 5.9
also shows the distributions of χ in various nucleic-acid structures.
5.3.4 Sugar/Glycosyl Combinations
To further illustrate conformational tendencies in polynucleotides, we gener-
ated adiabatic maps
6
for two models of deoxyadenosine in the {P, χ} space
(Figure 5.10) based on the CHARMM force field [415,803,804]. The first model
approximates solvation simply with a distance-dependent dielectric function. The
latter uses explicit representation of water molecules.
7
The adiabatic maps were calculated by dividing the {P, χ} grid to 3600
points, using 6
o
intervals for each angle. For each selected P , the values of
the 5 endocyclic sugar torsion angles were determined from eq. (5.1)using
τ
max
=40
o
. Starting structures were then generated from the set of variables
{τ
0
,τ
1
,τ
2
,τ
3
,τ
4
,χ} and minimized over the remaining degrees of freedom. Of
course, such a map only provides a reference for conformational flexibility, since
constraining (or freezing) the angles does not allow complete energy relaxation
over all the available degrees of freedom.
We note from Figure 5.10 essentially the same trends for the solvated
(top) and more approximate (bottom) models of four minima corresponding to
6
An adiabatic map is a simple way to examine molecular motion by characteristic low-energy
paths along a prescribed reaction coordinate (i.e., variations in specific conformational variables). For
each combination of these conformational coordinates, the entire potential energy of the system is
minimized to approximate behavior for the motion under study. Though simple in principle, specifi-
cation of the reaction coordinate is difficult in general, and the neglect of other degrees of freedom in
the process is clearly an approximation whose validity depends on the motion in question.
7
Namely, each nucleoside is enveloped in a water sphere of radius 11
˚
A, and the nonbonded inter-
actions are truncated at 12
˚
Ausinga2
˚
A buffer region, a potential shift function for the electrostatic
terms and a potential switch function for the van der Waals terms; see Chapter 10 for details of such
procedures.

5.3. Nucleic Acid Conformational Flexibility 149
60 120 180 240 300 360
60
120
180
240
300
360
χ
P
37
32
29
26
23
20
17
15
13
12
11
10
Energy (kcal/mol)
60 120 180 240 300 360
60
120
180
240
300
360
χ
P
31
27
23
20
17
15
13
11
10
9
8
Energy (kcal/mol)
Solvated dA
dA
Figure 5.10. Adiabatic maps for two models of deoxyadenosine in the {P, χ} space, the
sugar pseudorotation parameter and the sugar-to-base torsion angle, respectively, as calcu-
lated with the all-atom CHARMM 27 force field for nucleic acids [803, 809]. Hydration
is modeled with approximately 140 water molecules to produce the top figure, and ap-
proximated with a distance-dependent dielectric function to generate the bottom figure, as
described in the text. Only the internal energy of the dA system (i.e., excluding water/dA
and water/water energies) is used for plot generation in both cases.