Pecharsky V.K., Zavalij P.Y. Fundamentals of Powder Diffraction and Structural Characterization of Materials
Подождите немного. Документ загружается.

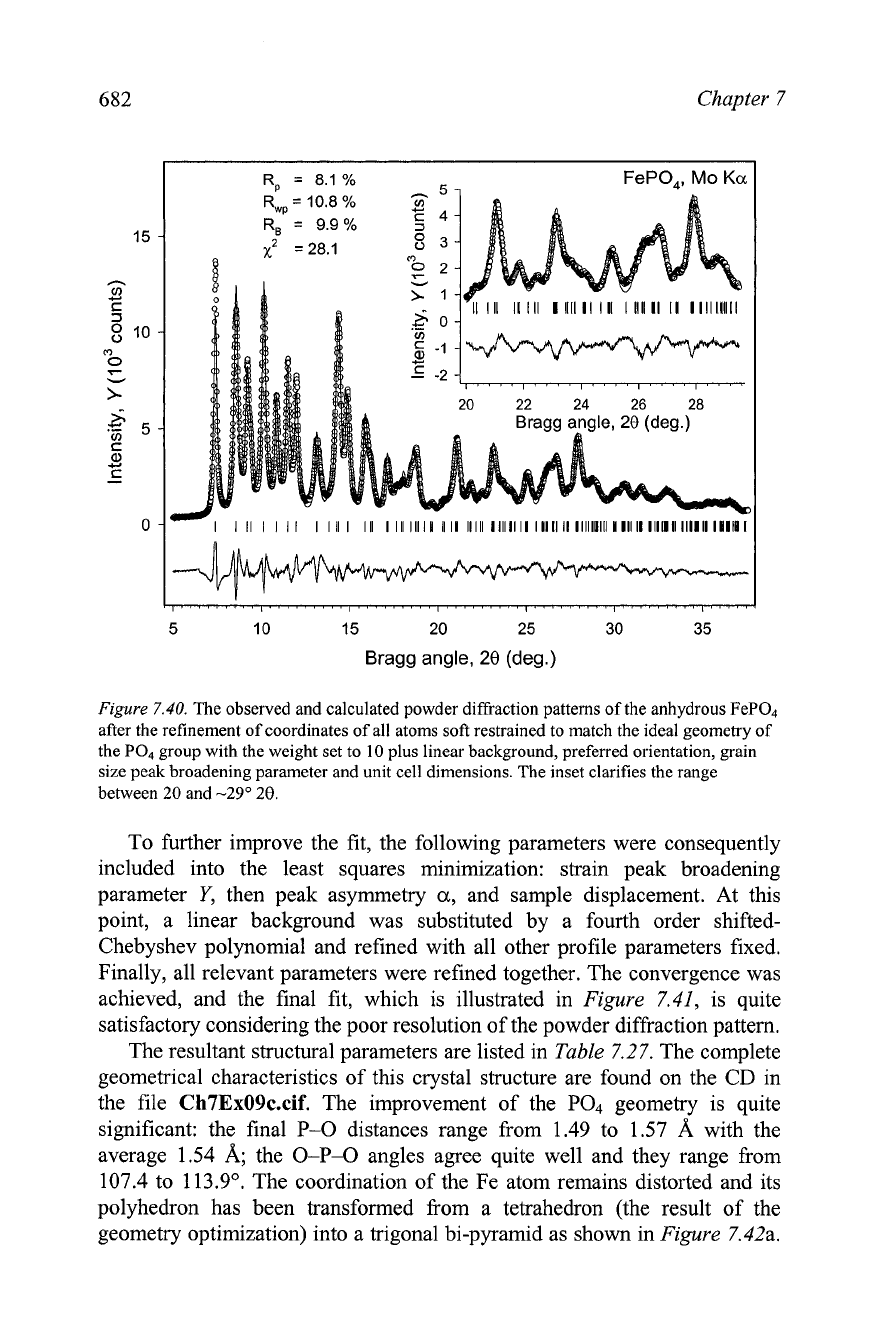
682
Chapter
7
5
10 15 20 25 30 35
Bragg angle,
28
(deg.)
Figure
7.40.
The observed and calculated powder diffraction patterns of the anhydrous FeP04
after the refinement of coordinates of all atoms soft restrained to match the ideal geometry of
the PO, group with the weight set to
10
plus linear background, preferred orientation, grain
size peak broadening parameter and unit cell dimensions. The inset clarifies the range
between
20
and
-29"
28.
To further improve the fit, the following parameters were consequently
included into the least squares minimization: strain peak broadening
parameter
Y,
then peak asymmetry
a,
and sample displacement. At this
point, a linear background was substituted by a fourth order shifted-
Chebyshev polynomial and refined with all other profile parameters fixed.
Finally, all relevant parameters were refined together. The convergence was
achieved, and the final fit, which is illustrated in
Figure
7.41,
is quite
satisfactory considering the poor resolution of the powder diffraction pattern.
The resultant structural parameters are listed in
Table
7.27.
The complete
geometrical characteristics of this crystal structure are found on the
CD
in
the file
Ch7Ex09c.cif.
The improvement of the
PO4
geometry is quite
significant: the final
P-0
distances range from 1.49 to 1.57
A
with the
average 1.54
A;
the
0-P-O
angles agree quite well and they range from
107.4 to 1 13.9O. The coordination of the Fe atom remains distorted and its
polyhedron has been transformed from a tetrahedron (the result of the
geometry optimization) into a trigonal bi-pyramid as shown in
Figure
7.42a.
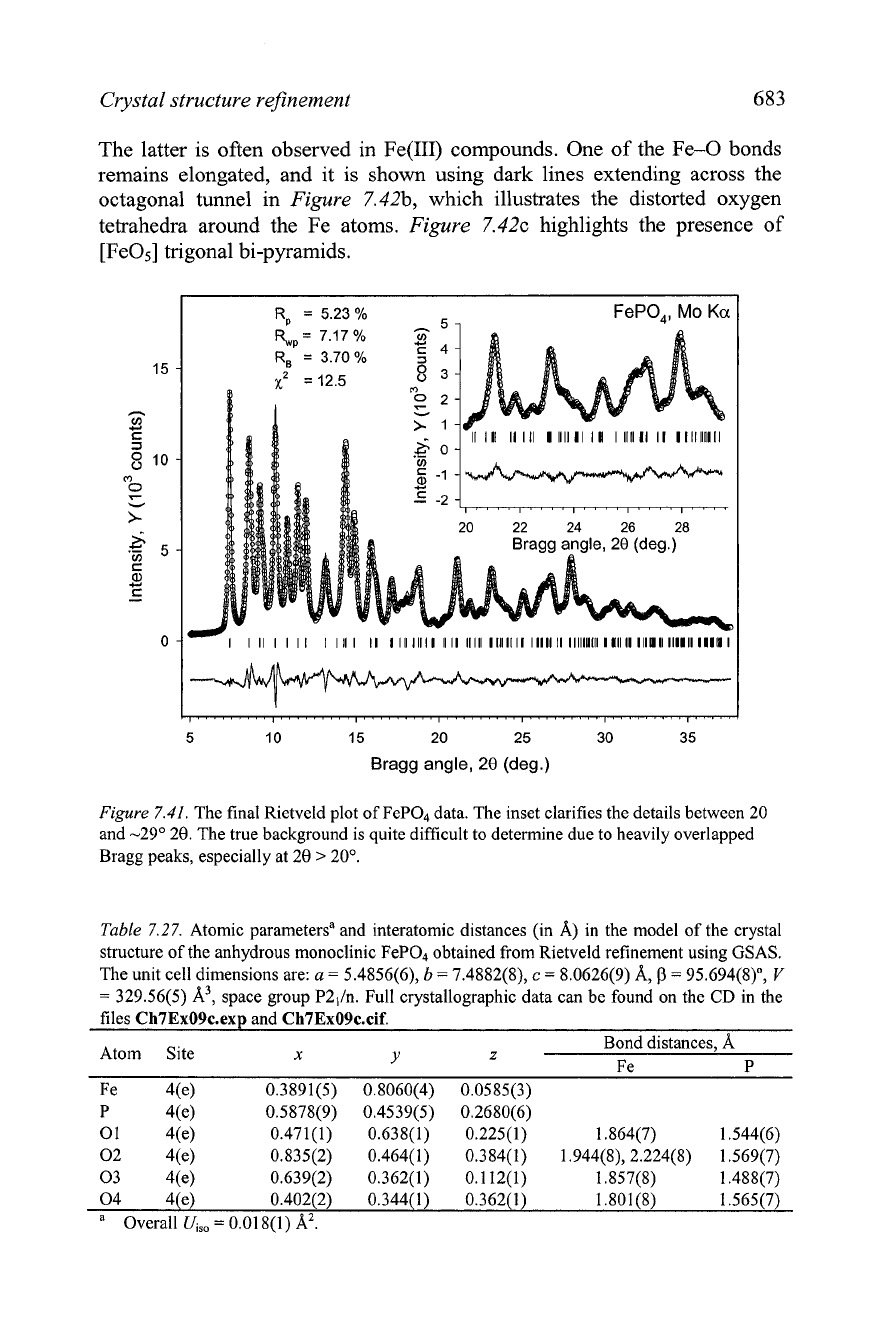
Crystal structure refinement
683
The latter is often observed in Fe(II1) compounds. One of the Fe-0 bonds
remains elongated, and it is shown using dark lines extending across the
octagonal tunnel in
Figure
7.42b, which illustrates the distorted oxygen
tetrahedra around the Fe atoms.
Figure
7.42~ highlights the presence of
[Fe05] trigonal bi-pyramids.
5 10 15
20
25
30 35
Bragg angle,
28
(deg.)
Figure
7.41.
The final Rietveld plot of FeP04 data. The inset clarifies the details between 20
and -29" 20. The true background is quite difficult to determine due to heavily overlapped
Bragg peaks, especially at 20
>
20'.
Table
7.27.
Atomic parametersa and interatomic distances (in A) in the model of the crystal
structure of the anhydrous monoclinic FeP04 obtained from Rietveld refinement using GSAS.
The unit cell dimensions are:
a
=
5.4856(6),
b
=
7.4882(8),
c
=
8.0626(9)
A,
0
=
95.694(8)",
V
=
329.56(5)
A3,
space group P2,In. Full crystallographic data can be found on the
CD
in the
files
Ch7Ex09c.e~~
and
Ch7Ex09c.cif.
Bond distances,
A
Atom Site
x
Y
z
Fe P
Fe 4(e) 0.3891(5)
0.8060(4) 0.0585(3)
P 4(e)
0.5878(9)
0.4539(5) 0.2680(6)
01 4(e)
0.471(1) 0.638(1)
0.225(1) 1.864(7) 1.544(6)
02 4(e)
0.835(2)
0.464(1) 0.384(1) 1.944(8), 2.224(8) 1.569(7)
03 4(e) 0.639(2)
0.362(1) 0.1 12(1) 1.857(8) 1.488(7)
04 4(e) 0.402(2)
0.344(1) 0.362(1) 1.801(8) 1.565(7)
a
Overall
Ui,,
=
0.01 8(1)
A'.
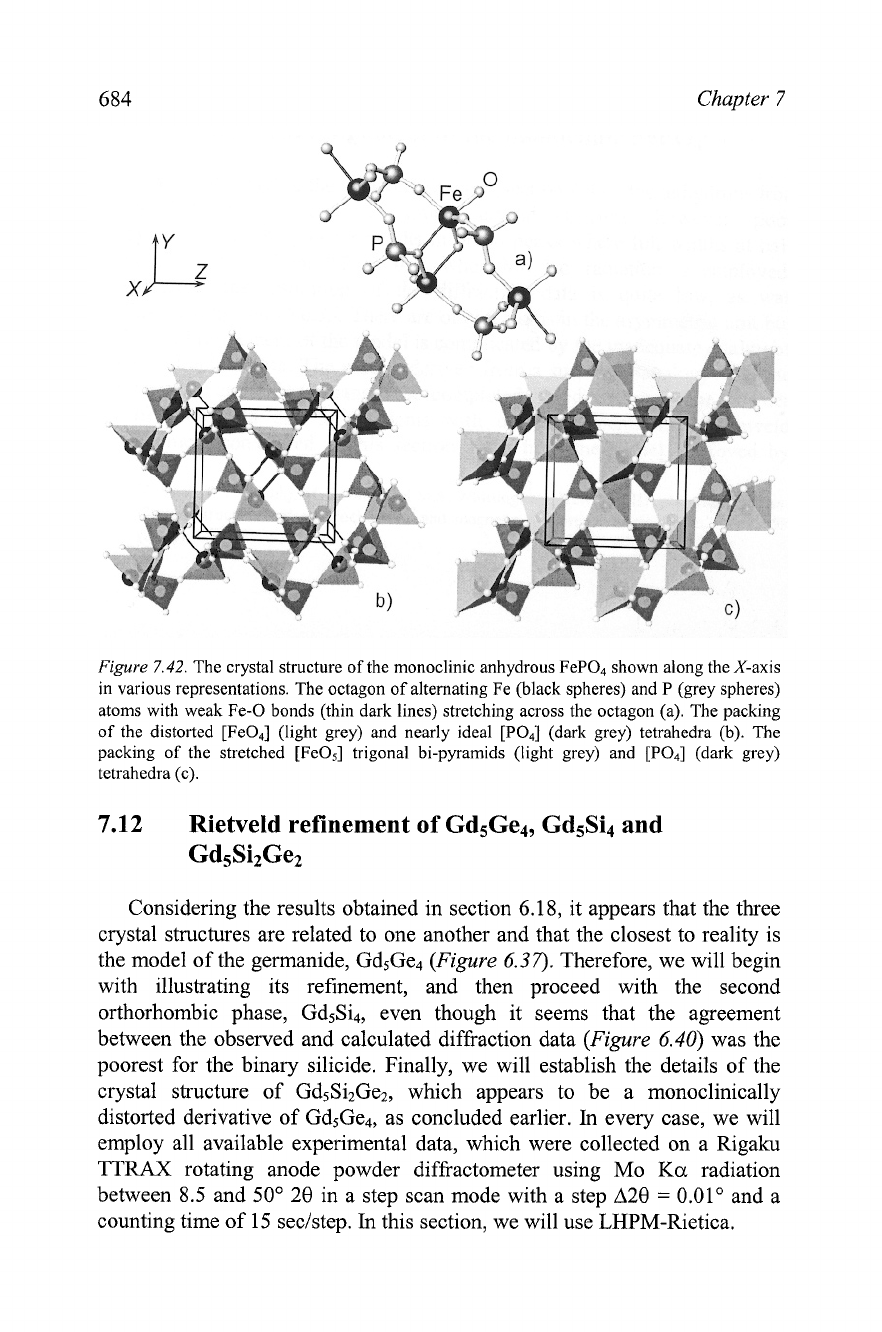
684
Chapter
7
Figure
7.42.
The crystal structure of the monoclinic anhydrous FeP04 shown along the X-axis
in various representations. The octagon of alternating Fe (black spheres) and P (grey spheres)
atoms with weak Fe-0 bonds (thin dark lines) stretching across the octagon (a). The packing
of the distorted [Fe04] (light grey) and nearly ideal [PO4] (dark grey) tetrahedra (b). The
packing of the stretched [FeOs] trigonal bi-pyramids (light grey) and [PO4] (dark grey)
tetrahedra (c).
7.12
Rietveld refinement
of
Gd5Ge4, Gd5Si4 and
Gd5Si2Ge2
Considering the results obtained in section 6.18, it appears that the three
crystal structures are related to one another and that the closest to reality is
the model of the germanide, Gd5Ge4
(Figure
6.37).
Therefore, we will begin
with illustrating its refinement, and then proceed with the second
orthorhombic phase, Gd5Si4, even though it seems that the agreement
between the observed and calculated diffraction data
(Figure
6.40)
was the
poorest for the binary silicide. Finally, we will establish the details of the
crystal structure of Gd5Si2Ge2, which appears to be a monoclinically
distorted derivative of Gd5Ge4, as concluded earlier.
In
every case, we will
employ all available experimental data, which were collected on a Rigaku
TTRAX
rotating anode powder diffiactometer using Mo
Ka
radiation
between 8.5 and 50" 20 in a step scan mode with a step A20
=
0.01" and a
counting time of 15 seclstep.
In
this section, we will use LHPM-Rietica.

Crystal structure rejinement
685
The coordinates of atoms taken from
Table
6.48
along with the unit cell
dimensions and all profile parameters determined from Le Bail's full pattern
decomposition are found in the input file, Ch7ExlOa.inp for LHPM-Rietica,
and the diffraction data are in the file
Ch7ExlO-MoKa.dat, located on the
CD.
The progress of Rietveld refinement is illustrated in
Table
7.28.
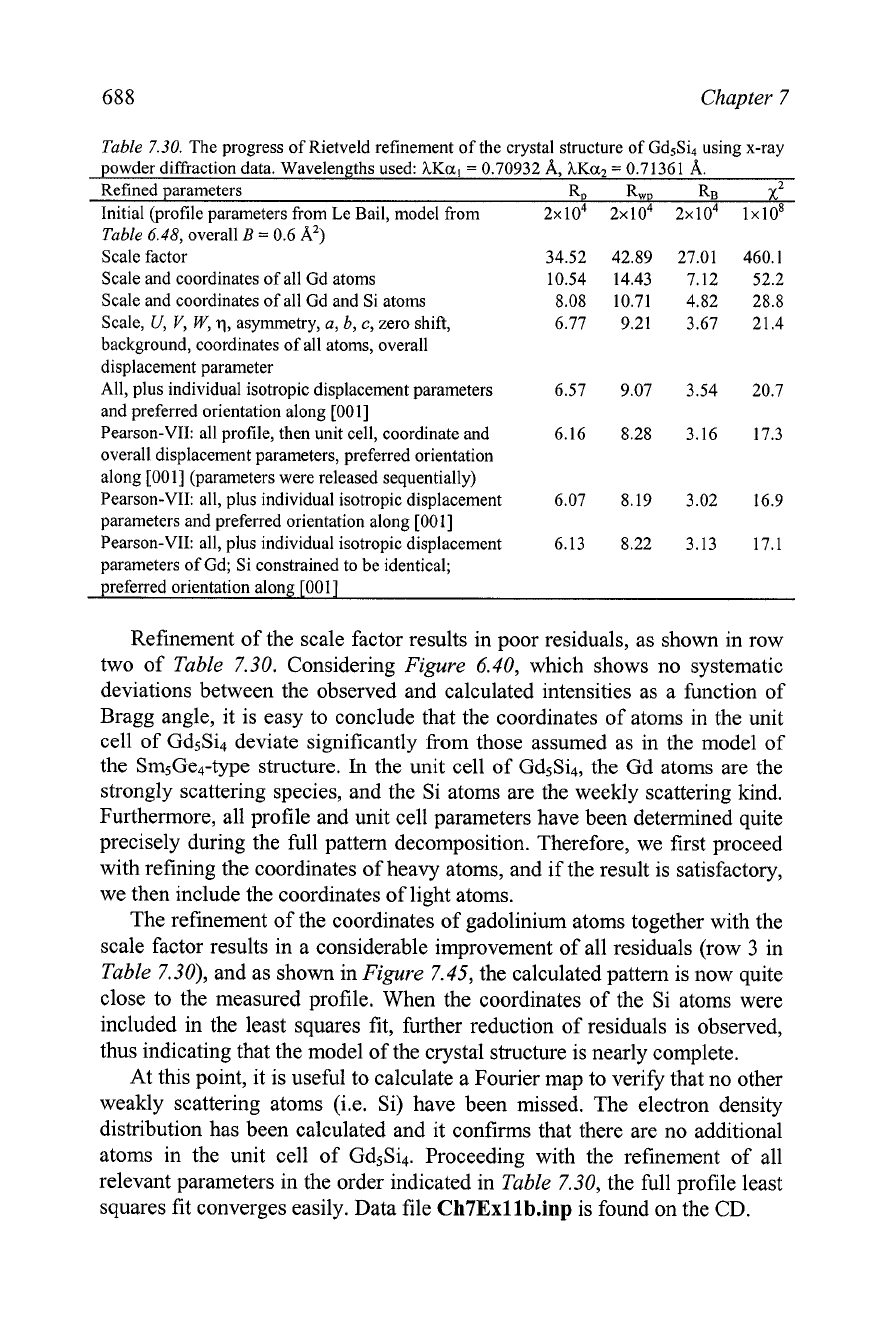
Table
7.28.
The progress of Rietveld refinement of the crystal structure of Gd5Ge4 using x-ray
powder diffraction data. Wavelengths used:
hKa,
=
0.70932 A, hKa2
=
0.71361 A.
Refined parameters RD RWD RB
x2
Initial (profile parameters from Le Bail, model from
3x10~ 3x104 3x104 2x10~
Table
6.48,
overall
B
=
0.6 A2)
Scale factor
11.54 14.65 9.53 43.17
Scale,
U,
V,
W,
q,
asymmetry,
a, b,
c,
sample
7.36 9.57 5.14 18.45
displacement, overall displacement parameter
All as above plus coordinates of all atoms and
5.85 7.90 3.38 12.62
background
All, plus individual isotropic displacement parameters
5.70 7.69 3.25 12.01
and preferred orientation along
[001Ia
a
The selection of the preferred orientation axis direction was based on the lowest residuals
after attempting to refine texture in the March-Dollase approximation along the three
major crystallographic axes.
Refinement quickly converges to low residuals, thus confirming the
correctness of the model of the crystal structure of this compound. Refined
individual parameters of all atoms are listed in
Table
7.29.
When individual
displacement parameters are refined in an anisotropic approximation,
residuals can be lowered further, but the displacement ellipsoid of the Gd3
atom becomes unphysical and therefore, the refinement was completed using
the isotropic approximation. Final values of all parameters can be found in
the data file Ch7ExlOb.inp on the
CD.
The observed and calculated powder
diffraction patterns are shown in
Figure
7.43.
Table
7.29.
Coordinates of atoms and individual isotropic displacement parameters in the
crystal structure of Gd5Ge4. The space group is Pnma. The unit cell dimensions are:
a
=
7.6997(3),
b
=
14.8309(4),
c
=
7.7861(3)
A.
All sites are fully occupied.
Atom Site
x
Y
z
B
(A2)
Gd
1 4(c) 0.2915(3) 1 14 -0.0008(3) 0.84(4)
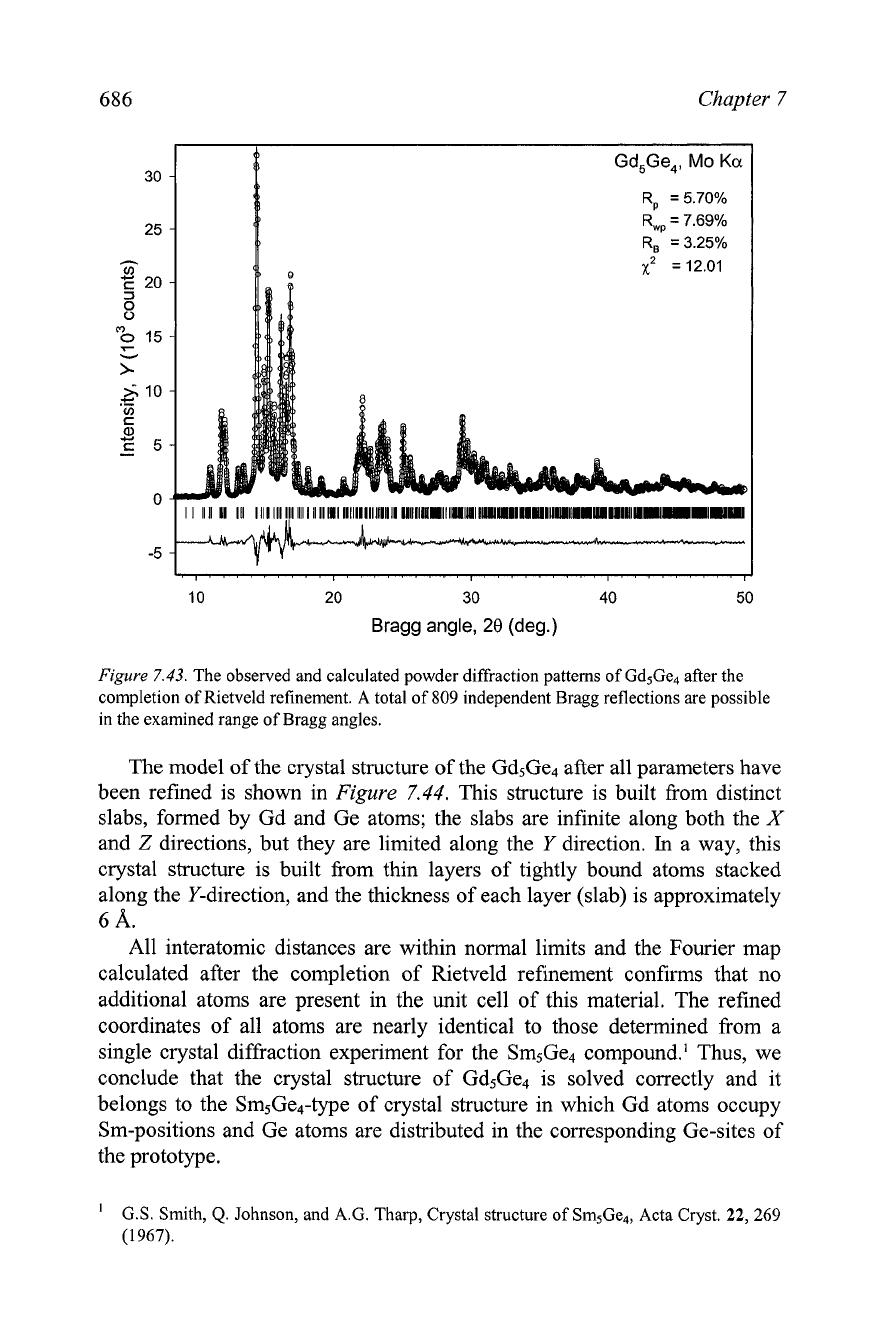
686
Chapter
7
10 20 30 40 50
Bragg angle,
28
(deg.)
Figure
7.43.
The observed and calculated powder diffraction patterns of Gd5Ge4 after the
completion of Rietveld refinement. A total of
809
independent Bragg reflections are possible
in the examined range of Bragg angles.
The model of the crystal structure of the Gd5Ge4 after all parameters have
been refined is shown in
Figure
7.44.
This structure is built from distinct
slabs, formed by Gd and Ge atoms; the slabs are infinite along both the
X
and
Z
directions, but they are limited along the
Y
direction.
In
a way, this
crystal structure is built from thin layers of tightly bound atoms stacked
along the Y-direction, and the thickness of each layer (slab) is approximately
6
A.
All interatomic distances are within normal limits and the Fourier map
calculated after the completion of Rietveld refinement confirms that no
additional atoms are present in the unit cell of this material. The refined
coordinates of all atoms are nearly identical to those determined from a
single crystal diffraction experiment for the Sm5Ge4 compound.' Thus, we
conclude that the crystal structure of Gd5Ge4 is solved correctly and it
belongs to the Sm5Ge4-type of crystal structure in which Gd atoms occupy
Sm-positions and Ge atoms are distributed in the corresponding Ge-sites of
the prototype.
'
G.S. Smith,
Q.
Johnson, and A.G. Tharp, Crystal structure of Sm5Ge4, Acta Cryst.
22,
269
(1967).
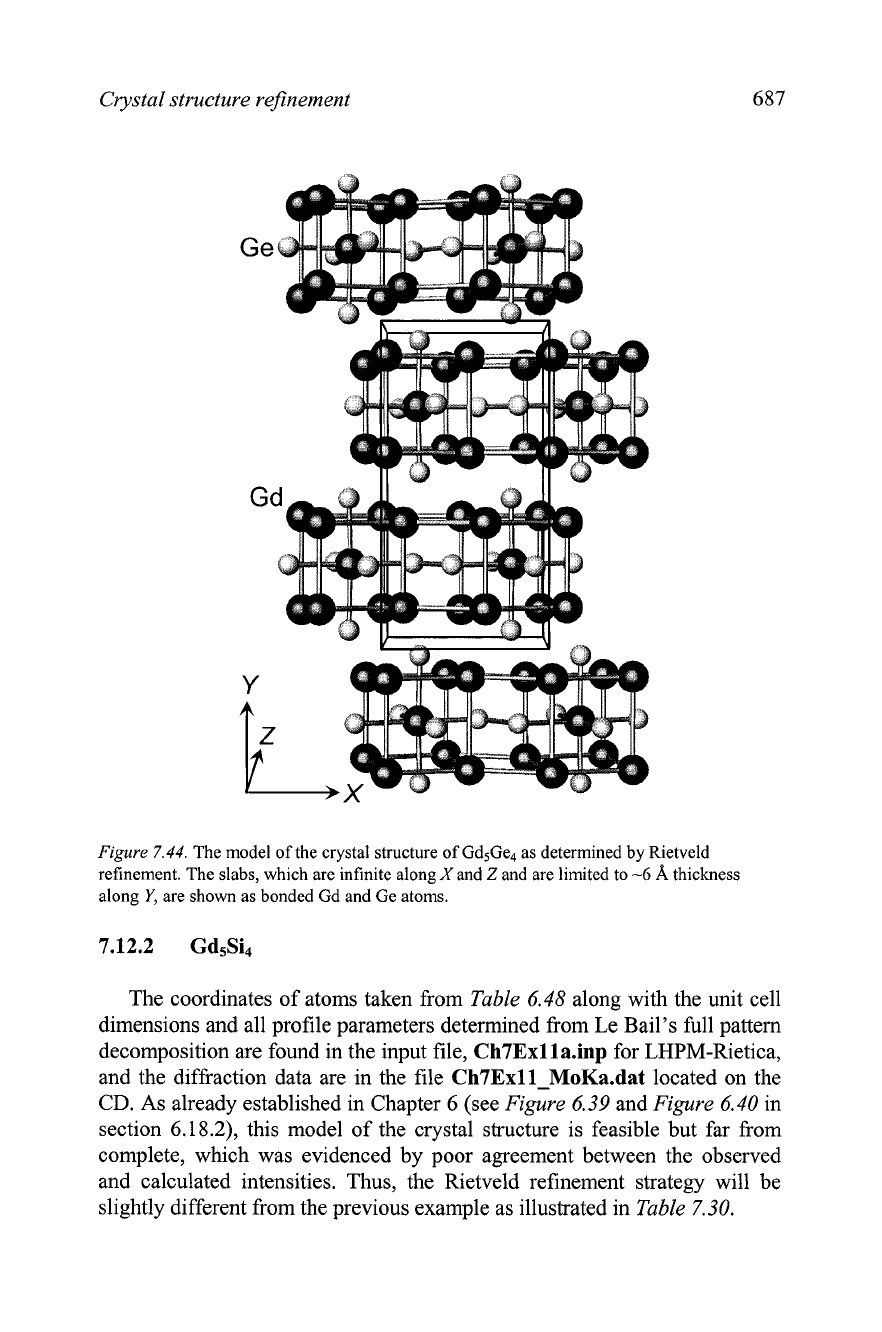
Crystal structure refinement
Figure
7.44.
The model of the crystal structure of Gd5Ge4 as determined by Rietveld
refinement. The slabs, which are infinite along
X
and
Z
and are limited to
-6
A
thickness
along
Y,
are shown as bonded Gd and Ge atoms.
The coordinates of atoms taken from
Table
6.48
along with the unit cell
dimensions and all profile parameters determined from Le Bail's full pattern
decomposition are found in the input file,
Ch7Exlla.inp
for LHPM-Rietica,
and the diffraction data are in the file
Ch7Exll-MoKa.dat
located on the
CD. As already established in Chapter 6 (see
Figure
6.39
and
Figure
6.40
in
section 6.18.2), this model of the crystal structure is feasible but far from
complete, which was evidenced by poor agreement between the observed
and calculated intensities. Thus, the Rietveld refinement strategy will be
slightly different from the previous example as illustrated in
Table
7.30.
688
Chapter
7
Table
7.30.
The progress of Rietveld refinement of the crystal structure of Gd5Si4 using x-ray
powder diffraction data. Wavelengths used:
hKal
=
0.70932
A,
hKaz
=
0.71361
A.
Refined parameters RD RwD RB
x2
Initial (profile parameters from Le Bail, model from
2~10~ 2~10~ 2~10~ lxlos
Table
6.48,
overall
B
=
0.6
A')
Scale factor
34.52 42.89 27.01 460.1
Scale and coordinates of all Gd atoms
10.54 14.43 7.12 52.2
Scale and coordinates of all Gd and Si atoms
8.08 10.71 4.82 28.8
Scale,
U,
V,
W,
q,
asymmetry,
a,
b,
c,
zero shift,
6.77 9.21 3.67 21.4
background, coordinates of all atoms, overall
displacement parameter
All, plus individual isotropic displacement parameters
6.57 9.07 3.54 20.7
and preferred orientation along
[001]
Pearson-VII: all profile, then unit cell, coordinate and
6.16 8.28 3.16 17.3
overall displacement parameters, preferred orientation
along
[001]
(parameters were released sequentially)
Pearson-VII: all, plus individual isotropic displacement
6.07 8.19 3.02 16.9
parameters and preferred orientation along
[OOl]
Pearson-VII: all, plus individual isotropic displacement
6.13 8.22 3.13 17.1
parameters of Gd; Si constrained to be identical;
preferred orientation along
[OOl]
Refinement of the scale factor results in poor residuals, as shown in row
two of
Table
7.30. Considering
Figure
6.40, which shows no systematic
deviations between the observed and calculated intensities as a function of
Bragg angle, it is easy to conclude that the coordinates of atoms in the unit
cell of
Gd5Si4 deviate significantly from those assumed as in the model of
the
Sm5Ge4-type structure.
In
the unit cell of Gd5Si4, the Gd atoms are the
strongly scattering species, and the Si atoms are the weekly scattering kind.
Furthermore, all profile and unit cell parameters have been determined quite
precisely during the full pattern decomposition. Therefore, we first proceed
with refining the coordinates of heavy atoms, and if the result is satisfactory,
we then include the coordinates of light atoms.
The refinement of the coordinates of gadolinium atoms together with the
scale factor results in a considerable improvement of all residuals (row
3
in
Table
7.30), and as shown in
Figure
7.45, the calculated pattern is now quite
close to the measured profile. When the coordinates of the Si atoms were
included in the least squares fit, further reduction of residuals is observed,
thus indicating that the model of the crystal structure is nearly complete.
At this point, it is useful to calculate a Fourier map to verify that no other
weakly scattering atoms (i.e. Si) have been missed. The electron density
distribution has been calculated and it confirms that there are no additional
atoms in the unit cell of Gd5Si4. Proceeding with the refinement of all
relevant parameters in the order indicated in
Table
7.30, the full profile least
squares fit converges easily. Data file
Ch7Exllb.h~
is found on the CD.
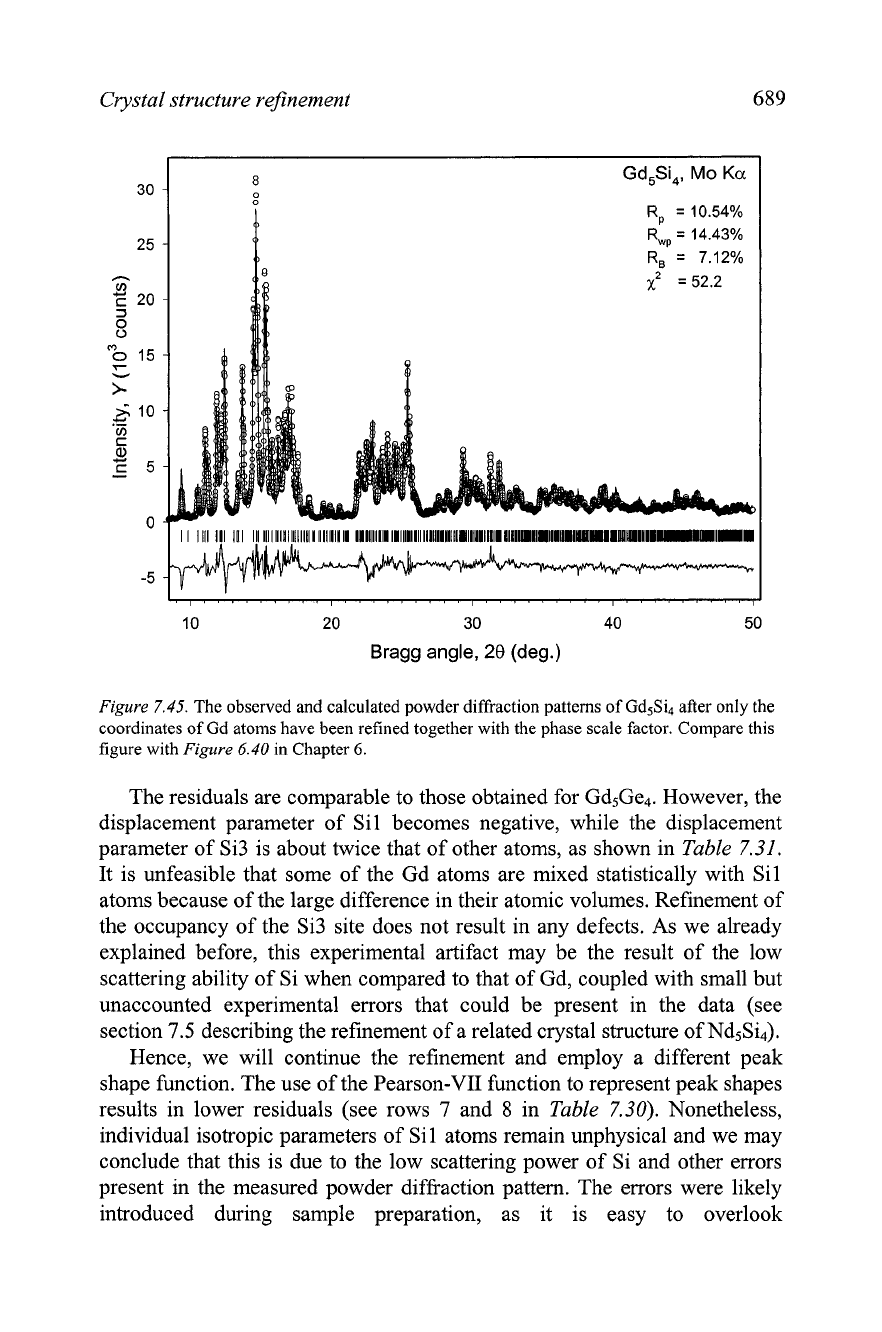
Crystal structure refinement
689
R,
=
10.54%
R,,
=
14.43%
10 20
30
40 50
Bragg angle,
28
(deg.)
Figure
7.45.
The observed and calculated powder diffraction patterns of Gd5Si after only the
coordinates of Gd atoms have been refined together with the phase scale factor. Compare this
figure with
Figure
6.40
in
Chapter
6.
The residuals are comparable to those obtained for Gd5Ge4. However, the
displacement parameter of Sil becomes negative, while the displacement
parameter of Si3 is about twice that of other atoms, as shown in
Table
7.31.
It is unfeasible that some of the Gd atoms are mixed statistically with Sil
atoms because of the large difference in their atomic volumes. Refinement of
the occupancy of the Si3 site does not result in any defects. As we already
explained before, this experimental artifact may be the result of the low
scattering ability of Si when compared to that of Gd, coupled with small but
unaccounted experimental errors that could be present in the data (see
section
7.5
describing the refinement of a related crystal structure of NdsSi4).
Hence, we will continue the refinement and employ a different peak
shape function. The use of the Pearson-VII function to represent peak shapes
results in lower residuals (see rows
7
and
8
in
Table
7.30).
Nonetheless,
individual isotropic parameters of Si1 atoms remain unphysical and we may
conclude that this is due to the low scattering power of Si and other errors
present in the measured powder diffraction pattern. The errors were likely
introduced during sample preparation, as it is easy to overlook
690
Chapter
7
inhomogeneities in the coverage of a flat sample holder with a powder when
the specimen has been prepared by dusting.
An
additional argument, which
supports the potential for problems with the specimen employed to collect
powder diffraction data, follows from considering the physical properties of
the silicide. According to Holtzberg
et
al.,' Gd5Si4 is ferromagnetic at room
temperature (its Curie temperature is
-335
K).
Thus, it is possible that an
unusual preferred orientation has been introduced into the powder sample,
even though the material is magnetically soft.
All things considered, the simplest solution is to refine the displacement
parameters of Si atoms in a pseudo-overall isotropic approximation by
constraining them to be identical, as is often done with light elements, such
as
C,
N, and
0
(see sections 7.7 to 7.11). The resulting residuals (the last
row in Table
7.30)
are only slightly higher when compared to the refinement
of the individual displacement parameters of all atoms. The final free
variables in the crystal structure of Gd5Si4 are listed in Table
7.32.
A small
difference in the unit cell dimensions obtained using different peak shape
functions is normal because peak asymmetry has been treated differently.
The observed and calculated powder diffraction patterns after all parameters
in the crystal structure of
Gd5Si4 have been refined are shown in
Figure
7.46.
Table
7.31.
Coordinates of atoms and individual isotropic displacement parameters in the
crystal structure of Gd5Si4 fully refined by Rietveld technique using the pseudo-Voigt peak
shape function. The space group is Pnma. The unit cell dimensions are:
a
=
7.4896(4),
b
=
14.7544(8),
c
=
7.7519(4)
A.
All sites are fully occupied.
Atom Site
x
Y
z
B
(A2)
Gd
1
4(c) 0.3539(4) 114 0.0099(3) 0.89(5)
Gd2 8(d) 0.0294(2) 0.0974(1) 0.1787(2) 1.18(3)
Gd3 8(d) 0.3 159(2) 0.8780(1) 0.1837(2) 0.98(3)
Ge 1 4(c) 0.244(1) 1 14 0.367(1) -0.6(2)
Ge2 4(c) 0.990(2) 1 /4 0.919(2) 1.0(3)
Ge3 8(d) 0.159(1) 0.9510(6) 0.490(1) 2.2(2)
Table
7.32.
Coordinates of atoms and individual isotropic displacement parameters in the
crystal structure of Gd5Si4 fully refined by Rietveld technique using the Pearson-VII peak
shape function. The space group is Pnma. The unit cell dimensions are:
a
=
7.4877(2),
b
=
14.7496(5),
c
=
7.7497(2) A. All sites are fully occupied.
Atom Site
x
Y
z
B
(A2)
Gd 1 4(c) 0.3544(2) 1 /4 0.0102(3) 0.88(4)
Gd2 8(d) 0.0289(2) 0.0987(1) 0.1790(2) 1.14(3)
Gd3 8(d) 0.3 159(2) 0.8778(1) 0.1839(2) 0.94(3)
Ge
1
4(c) 0.246(1) 1 /4 0.369(1) 0.9(1)
'
F.
Holtzberg,
R.J.
Gambino, and
T.R.
McGuire, New ferromagnetic 5:4 compounds in the
rare earth silicon and germanium systems,
J.
Phys. Chem. Solids
28,
2283 (1967).
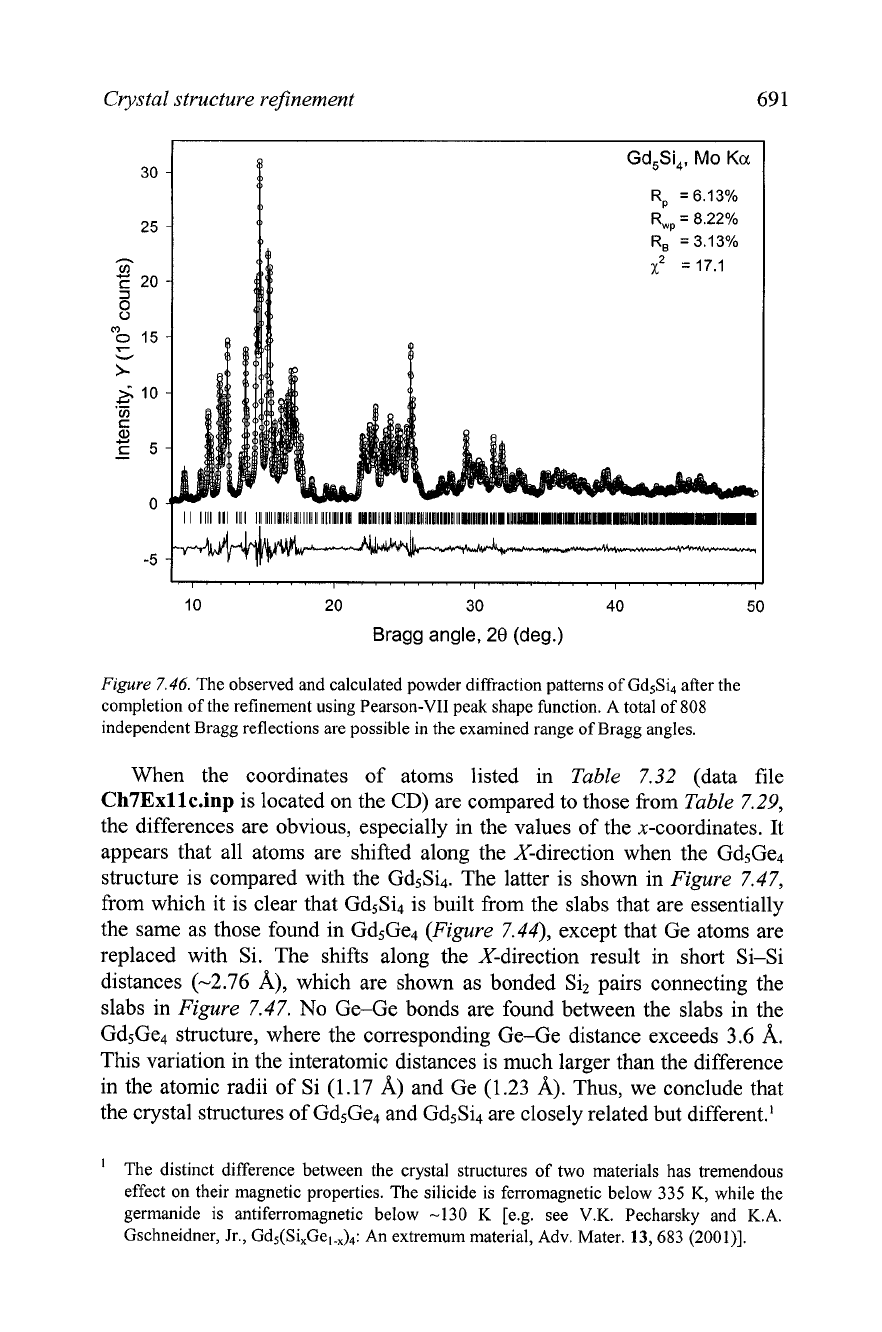
Crystal structure refinement 69 1
10
20
30
40
50
Bragg angle,
28
(deg.)
Figure
7.46.
The observed and calculated powder diffraction patterns of Gd5Si4 after the
completion of the refinement using Pearson-VII peak shape function. A total of
808
independent Bragg reflections are possible in the examined range of Bragg angles.
When the coordinates of atoms listed in Table 7.32 (data file
Ch7Exllc.inp
is located on the
CD)
are compared to those fkom Table 7.29,
the differences are obvious, especially in the values of the x-coordinates. It
appears that all atoms are shifted along the X-direction when the Gd5Ge4
structure is compared with the Gd5Si4. The latter is shown in Figure 7.47,
from which it is clear that Gd5Si4 is built from the slabs that are essentially
the same as those found in Gd5Ge4 (Figure 7.44), except that Ge atoms are
replaced with Si. The shifts along the X-direction result in short Si-Si
distances (-2.76
A), which are shown as bonded Si2 pairs connecting the
slabs in Figure 7.47. No Ge-Ge bonds are found between the slabs in the
Gd5Ge4 structure, where the corresponding Ge-Ge distance exceeds
3.6
A.
This variation in the interatomic distances is much larger than the difference
in the atomic radii of Si (1.17 A) and Ge (1.23
A).
Thus, we conclude that
the crystal structures of Gd5Ge4 and Gd5Si4 are closely related but different.'
The distinct difference between the crystal structures of two materials has tremendous
effect on their magnetic properties. The silicide is ferromagnetic below 335 K, while the
germanide is antiferromagnetic below -130 K
[e.g, see V.K. Pecharsky and K.A.
Gschneidner, Jr.,
Gd5(Si,Gel.,),: An extremum material, Adv. Mater.
13,
683
(2001)l.