Pecharsky V.K., Zavalij P.Y. Fundamentals of Powder Diffraction and Structural Characterization of Materials
Подождите немного. Документ загружается.

672
Chapter
7
fit, yet another 3% reduction has been achieved. All individual isotropic
displacement parameters are positively defined but
Ui,,
of Mn2,
V2
and
06
were noticeably higher and increase continually, which points to possible
vacancies in these positions.'
The deficiency, if any, makes sense
fkom a structural point of view as
well, because the suspected defect sites are located along the tunnels, which
exist around the 6-fold and 3-fold axes, thus leaving the main framework
intact. At this point, the overall
U,,,
were assigned separately to all metal and
oxygen atoms and the occupancies of the three suspicious sites were refined
(Figure
7.34).
20 30 40 50 60 70 80 90 100 110 120 130
Bragg angle,
20
(deg.)
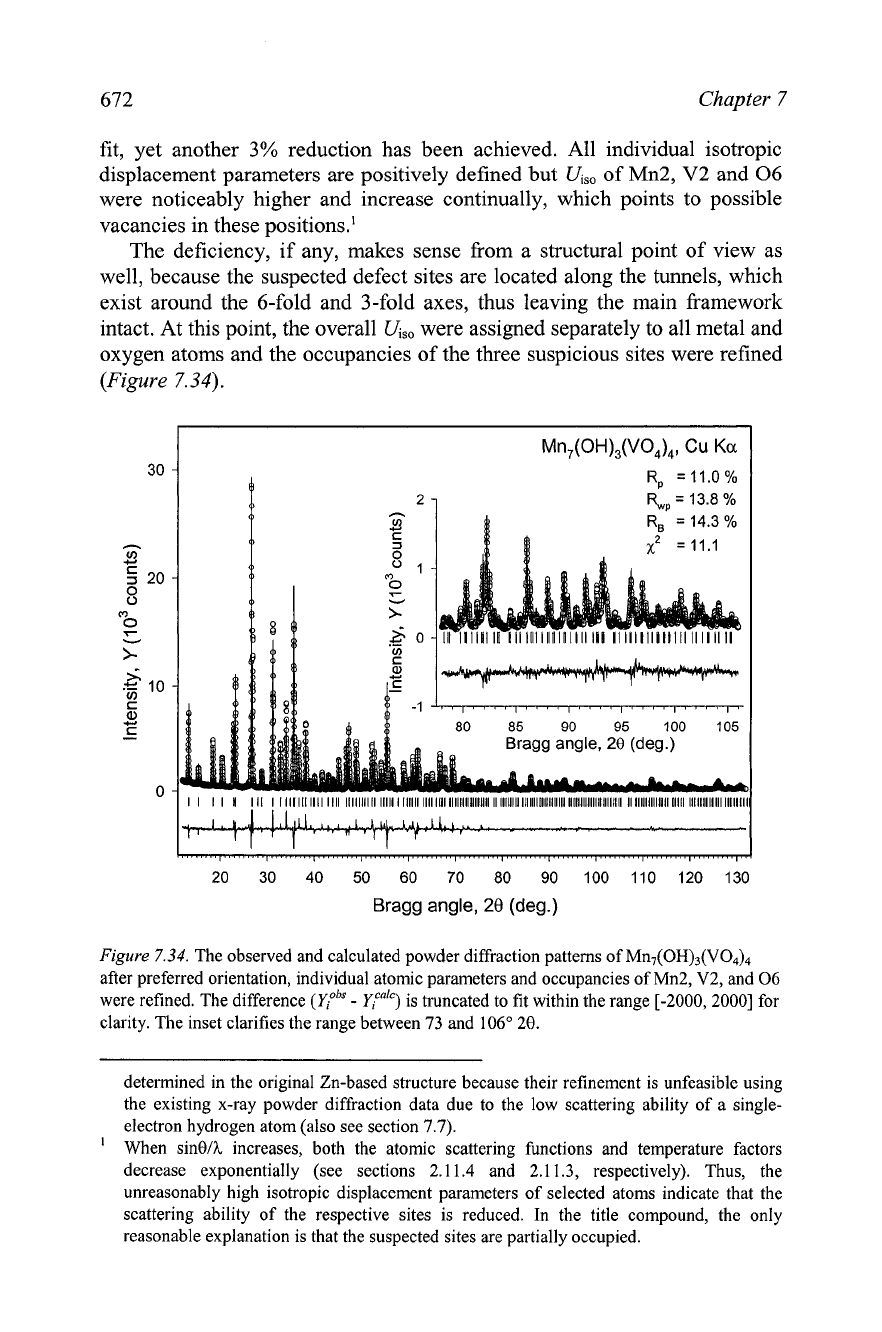
Figure
7.34.
The observed and calculated powder diffraction patterns of MII,(OH)~(VO~)~
after preferred orientation, individual atomic parameters and occupancies of Mn2, V2, and 06
were refined. The difference
(cobs
-
Ca")
is truncated to fit within the range [-2000,2000] for
clarity. The inset clarifies the range between 73 and 106" 20.
determined in the original Zn-based structure because their refinement is unfeasible using
the existing x-ray powder diffraction data due to the low scattering ability of a single-
electron hydrogen atom (also see section 7.7).
'
When sinO/h increases, both the atomic scattering functions and temperature factors
decrease exponentially (see sections 2.11.4 and 2.11.3, respectively). Thus, the
unreasonably high isotropic displacement parameters of selected atoms indicate that the
scattering ability of the respective sites is reduced. In the title compound, the only
reasonable explanation is that the suspected sites are partially occupied.

Crystal structure refinement
673
A difference Fourier map, calculated at this point, reveals an additional
small electron density maximum in the tetrahedral cavity next to the partially
occupied V2.' Thus, it is reasonable to assume that the V2 site splits into two
independent partially occupied positions with the coordinates, which
distribute V atoms in a random fashion in two adjacent tetrahedral positions
rather than being simply vanadium-deficient. We label these two sites as
V2a (corresponding to the former V2) and V2b (corresponding to the Fourier
peak). Refinement of this model slightly improves the fit. Subsequently,
additional profile parameters (Y,
Yo, and sample displacement) were included
in the refinement, followed by a typical procedure of refining the porosity in
the Suortti approximation with fixed atomic coordinates and Ui,,, and then
fixing the porosity parameters for the remainder of the refinement.
Obviously, the prohibitively short distance between V2a
and V2b
mandates that the following relationship holds:
Furthermore, the analysis of the values of the occupation factors refined for
V2a, V2b, and 06 point to the following relationship:
These two relationships can be easily programmed in GSAS and in the
majority of Rietveld software codes by using a constraint apparatus, which
was briefly discussed above (see section 7.3.3 and
Eq.
7.9).
Since the
constraints affect only the shifts that are determined during every least
squares refinement cycle but not the values of the related parameters, the
latter should be synchronized manually prior to imposing constraints. For
example, in our case when the computed shift for
gv2a
is 0.02, then the new
values of the constrained parameters (Eqs. 7.9, 7.11 and 7.12) will be
calculated as follows:
If before the beginning of the constrained refinement, gvza is not equal to
go6, e.g. they are 0.6 and 0.8, respectively, then after adding the shifts
according to Eq. 7.13, the corresponding values will become 0.62 and 0.82.
Thus, if needed, parameters constrained in this way should be matched
'
This peak is characterized by a reasonable tetrahedral configuration created by the oxygen
atoms, except that it is too close to the existing
V2.
Considering the deficiency of the
V2
site, it is feasible that the structure contains two types of
[V04]
tetrahedra distributed
in
the
structure in a random fashion.

674
Chapter
7
manually: in this example, both
gv2a
and go6 should be set to identical values
and
gvza
and
gv2b
should sum up to unity.'
The relationships between the occupancies in this crystal structure have
both the chemical and physical sense. The V2 atom and the surrounding 4
oxygen atoms (three 04 and one 06) in a fully ordered structure create a
chain shown in
Figure
7.35a, where the occupied (grey) and empty (white)
tetrahedra are alternated. Vanadium atoms can also occupy pairs of corner-
sharing tetrahedra, thus forming a well-known V2O7 group (hatched in
Figure
7.35b). When a "mistake" occurs and a vanadium atom "jumps" to
the next empty site, the corner sharing pair of empty tetrahedra appears, in
which the shared corner must be vacated because the corresponding oxygen
atom is no longer bound to any vanadium atom. Therefore, vacancies on the
06 sites, constrained as shown in Eq. 7.12, should exist, as was confirmed
by the independent refinement of
go6.
The subsequent refinement included profile parameters X, Y, Xu, Y,, peak
asymmetry, sample displacement and transparency shift. Preferred
orientation was switched from the March-Dollase to the 8th-order spherical
harmonics expansion
(6
variables total) and 12 coefficients of the shifted-
Chebyshev polynomial background approximation were employed. A
reasonably good fit, shown in
Figure
7.36, was achieved as a result.
-
V,O,
-
vacant site
Figure
7.35.
Chains of tetrahedra in the MQ(OH)~(VO~)~ structure. The fully ordered chain
(a) with the composition V04, where vanadium occupies every other tetrahedron (shaded) and
the remaining are vacant (white). The disordered chain
(b),
where some vanadium atoms
occupy pairs of neighboring tetrahedra, forming V2O7 groups (hatched), thus creating
vacancies on the 06 site and resulting in the chemical composition
(V04)1-x(V207)xiZ
Another example of synchronizing constrained parameters can be given considering the
coordinates of
V1 and 02
-
05 atoms in this structure. All of them are located in the 6(c)
sites, with the coordinates of the independent atom
xk.
Hence, when entering the
respective coordinate parameters it is necessary to ensure that
y
=
-x.
Crystal structure rejkement
675
Mn,(OH),(VO,),,
Cu
Ka
R,
=
9.0
%
R,,=
11.7
%
R,
=
10.7
%
0
85
90 95 100 105
Bragg angle,
20
(deg.)
I I I
I
I
U
II
1
I IIIIII
1111
1111
1111l11111 IIIII
I
IIIIII
1111
1111
1111111111111111
11
1111111111111111111lllllllllllllllllllllllllllll
111111111111111
11111
111111111111111111
20 30 40 50 60 70 80 90 100 110 120 130
Bragg angle, 20 (deg.)
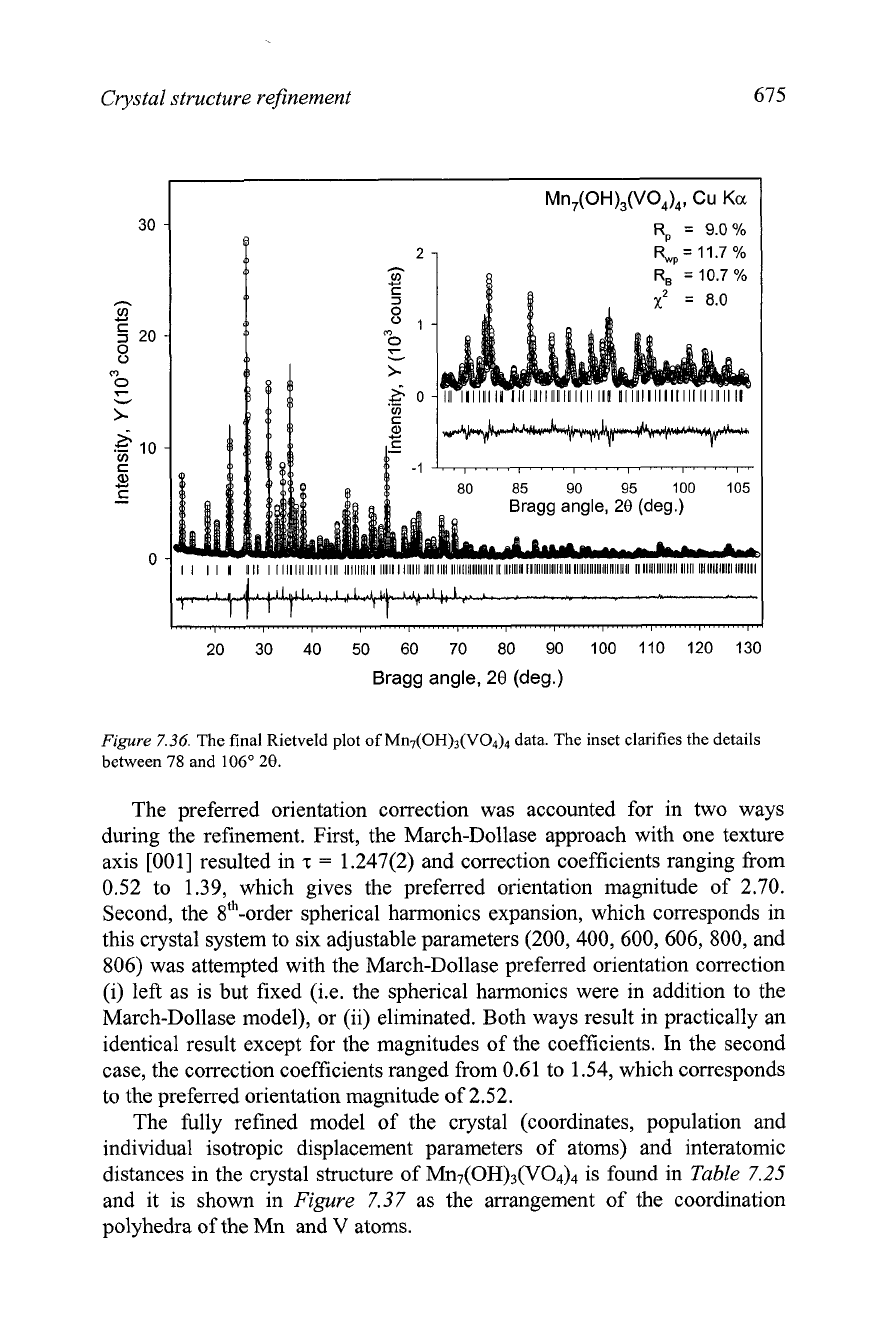
Figure
7.36.
The final Rietveld plot of MII~(OH)~(VO~)~ data. The inset clarifies the details
between
78
and
106"
20.
The preferred orientation correction was accounted for in two ways
during the refinement. First, the March-Dollase approach with one texture
axis
[001] resulted in
z
=
1.247(2) and correction coefficients ranging from
0.52 to
1.39,
which gives the preferred orientation magnitude of 2.70.
Second, the sth-order spherical harmonics expansion, which corresponds in
this crystal system to six adjustable parameters (200,400, 600, 606, 800, and
806) was attempted with the March-Dollase preferred orientation correction
(i) left as is but fixed
(i.e. the spherical harmonics were in addition to the
March-Dollase model), or (ii) eliminated. Both ways result in practically an
identical result except for the magnitudes of the coefficients.
In
the second
case, the correction coefficients ranged from 0.61 to 1.54, which corresponds
to the preferred orientation magnitude of
2.52.
The fully refined model of the crystal (coordinates, population and
individual isotropic displacement parameters of atoms) and interatomic
distances in the crystal structure of MQ(OH)~(VO~)~ is found in
Table
7.25
and it is shown in
Figure
7.37
as the arrangement of the coordination
polyhedra of the Mn and
V
atoms.

676
Chapter
7
Table
7.25.
The final atomic parametersa9b and interatomic distances (in A) in the crystal
structure of
M~I~(OH)~(VO~)~ obtained from Rietveld refinement using GSAS. The fully
refined unit cell parameters are:
a
=
13.2292(1),
c
=
5.25467(6) A,
V
=
796.42(2) A3, the
space group is P63mc. Full set of crystallographic data is also found on the
CD
in the files
Ch7ExOSc.exp and Ch7ExOSc.cif.
Bond distances,
A
Atom Site
x
Y
z
Mn 1 Mn2 V1 V2a
1.66 1 (6)3
05 6~ 0.9199(3) -0.9199(3) 0.586(2) 2.214(7),, 1.637(6)
2.302(8),
06 2b 113 213 0.118(3) 1.81(2)
~1~ 6c 0.433 -0.433 0.156
a
Ui,,
is 0.0145(2)
AZ
for metal atoms and 0.0140(8)
A2
for
0
atoms.
The population parameters are as follows: gv2,
=
go,
=
0.819(4), gv2b
=
0.181(4)
=
1
-
gvza, g~,,2
=
0.873(4)
s
(1-2/3gv2b). All other sites are fully occupied.
This parameter was kept constant at all times to maintain a fixed origin of coordinates
along the Z-axis. Although GSAS enables automatic fixation of the origin of coordinates,
the refinement may become unstable, especially when the fit is far from the best.
Taken from the Zn-based compound and kept constant during the refinement.
In
addition to the already discussed vacancy relationships among V2a,
V2b and 06 atoms, the refined deficiency of the Mn2 site is nearly equal to
213 of the vacancies observed on the V2b site. Although the experimentally
established relationship is approximate, when obeyed exactly
(i.e. when g,,,
=
1
-
2/3gV2+,), the oxidation state of manganese in the Mn2 site is
3+,
while
it is 2+ for the Mnl site. All things considered, the following expression
describes the chemical composition of M~I~(OH)~(VO~)~:
This formula has been confirmed from single crystal diffraction data
(RF
=
2.2
%),
which give x
=
0.202(3).' The population of the Mn2 site is
0.792(4), slightly lower than expected 1
-
213x
=
0.865.
A
small difference is
acceptable because a tiny single crystal may not be representative of a large
polycrystalline sample, especially in the case of occupational disorder.
P. Zavalij,
S.
Luta, and
M.S.
Whittingham, unpublished.

Crystal structure refinement
Figure
7.37.
The model of the crystal structure of
Mn7~y(OH)3(V04)4-,(V207)x12
shown along
the Z-axis. The Mnl sites (~n~') are shown as light spheres inside the dark grey [MnOs]
octahedra, the partially occupied Mn2 sites (~n~') are shown as light spheres in the light grey
[MnO,] octahedra, both the
V1
and
V2
sites are shown as dark spheres inside the light grey
[V04]
tetrahedra. Hydrogen atoms from the hydroxyl groups are not shown in this figure.
7.11
Rietveld refinement of the monoclinic FeP041
As established in the previous chapter (section 6.17), the anhydrous iron
phosphate has a relatively simple crystal structure, however, poor
crystallinity of the powder results in broad peaks where full widths at half
maximum vary from 0.2 to
0.5'
when Mo
Ka
radiation is employed.
Therefore, the resolution of the diffraction data is quite low, as was
illustrated in
Figure
6.33.
There are only
6
atoms in the asymmetric unit but
Rietveld refinement of the model is complicated by the inadequate quality of
the diffraction data. The model, derived from a suspected analogy with the
hydrated FeP04.2H20, cannot be completed based solely on the powder
diffraction data due to problems with the experiment. Thus, Rietveld
refinement considered in this section starts from the model improved by
'
Y.
Song, P.Y. Zavalij, M. Suzuki,
and
M.S. Whittingharn, New iron(II1) phosphate phases:
Crystal structure and electrochemical and magnetic properties, Inorg. Chem.
41,
5778
(2002).

678 Chapter
7
geometry optimization using quantum chemical minimization of
approximate coordinates of atoms, as was discussed in the previous chapter.
Furthermore, data quality precludes an unrestrained refinement of even the
optimized model, and similar to the example considered in section 7.8, soft
restraints should be imposed on the geometry of the
PO4 groups. We will,
therefore, take this opportunity to illustrate the role of soft restraint
weighting in Rietveld refinement.
The experimental powder diffraction pattern was collected on a rotating
anode Rigaku
TTRAX powder diffractometer using monochromatized Mo
Ka
radiation from 5 to 50'
28
in a step scan mode with a 0.01' step and
counting time of 10 seclstep. The following parameters were employed at
the beginning of this refinement:
-
The initial structure model, derived and optimized in the previous chapter
(Table
6.47).
-
The default profile parameters from the instrumental parameter file
Rigaku.prm obtained from the refinement of the La& standard as
described in section 7.6.
-
The sample shift parameter
S,
=
3.99 for the sample displacement s
=
-0.087 mm obtained together with the unit cell dimensions at a stage of
lattice parameters refinement.
-
The space group P2Jn and the unit cell dimensions
a
=
5.489, b
=
7.493,
c
=
8.055 A, and
P
=
95.81•‹, determined earlier.
-
The overall isotropic displacement parameter
U,,,
=
0.015 A2;
The initial model with all needed crystallographic parameters is found on
the
CD
in the files Ch7Ex09a.e~~ and Ch7Ex09a.cif, and the experimental
data are in the file Ch7Ex09-MoKa.raw. Only the range from 5 to 37.6' 28
was used in all calculations.' The background was approximated manually
with a straight line (i.e. two coefficients of a shifted-Chebyshev polynomial
were employed in this approximation) since unambiguous automatic
background determination was impossible at the beginning of the refinement
due to heavily overlapped Bragg peaks. Only two data points (the first and
the last in the range) were used for background estimation. The initial
refinement of the phase scale factor resulted in
R,
=
62.7% (Table
7.26)
and
a quite poor fit, which showed that all calculated Bragg peaks were too
narrow. Therefore, both peak broadening parameters,
X
and
Y,
were
manually increased by a factor of
10,
yielding an acceptable weighted profile
residual of 35.6% and resulting in a tolerable fit as shown in Figure
7.38.
'
This range of Bragg angles corresponds to a
20,,,
2
89'
when using Cu
Ka
radiation. The
use of Mo
Ka
radiation in this case was justified by a large goniometer radius
(285
rnm)
and therefore, potentially high resolution, and by the presence of the significant amount of
Fe in the material (iron strongly absorbs Cu
Ka
radiation, see
Table
2.3,
and creates a
substantial fluorescent background).
Crystal structure
refinement
679
Table 7.26.
The progress of Rietveld refinement of the crystal structure of anhydrous FeP04
using low resolution x-ray powder diffraction data.
Refined parameters RD
Rw~
R~
x2
Initial
(CD:
Ch7Ex09a.e~~
and
Ch7Ex09a.N)
75.2 77.2 98.6 1414.
Scale factor, linear background adjusted manually
57.4 62.7 67.4 933.
Peak broadening
X
and
Y
multiplied by
10
then phase scale
29.4 35.6 32.6 301.
(Figure 7.38)
Plus two background coefficients, coordinates, PO4 group soft-
13.3 16.8 22.6 67.6
restrained with weight
4
Same but soft-restraints with weight increased to
10
11.1
13.8 13.8 46.2
Plus grain size broadening,
X
8.5 11.1 10.7 30.1
Plus unit cell dimensions,
PO
[OlO],
overall
Ui,,
(Figure 7.40)
8.1 10.8 9.9 28.1
(CD:
Ch7Ex09b.e~~
and
Ch7ExO9b.cif)
Plus strain broadening,
Y,
then asymmetry and sample
7.3 9.7 6.2 22.7
displacement
Four coefficients of the background
6.5 8.6 4.1 17.9
X,
Y,
peak asymmetry,
X,,
Y,;
(Figure 7.41, Table 7.27) (CD:
5.2 7.1 3.6 12.3
Ch7Ex09c.e~~
and
Ch7Ex09c.cif)
Bragg angle,
28
(deg.)
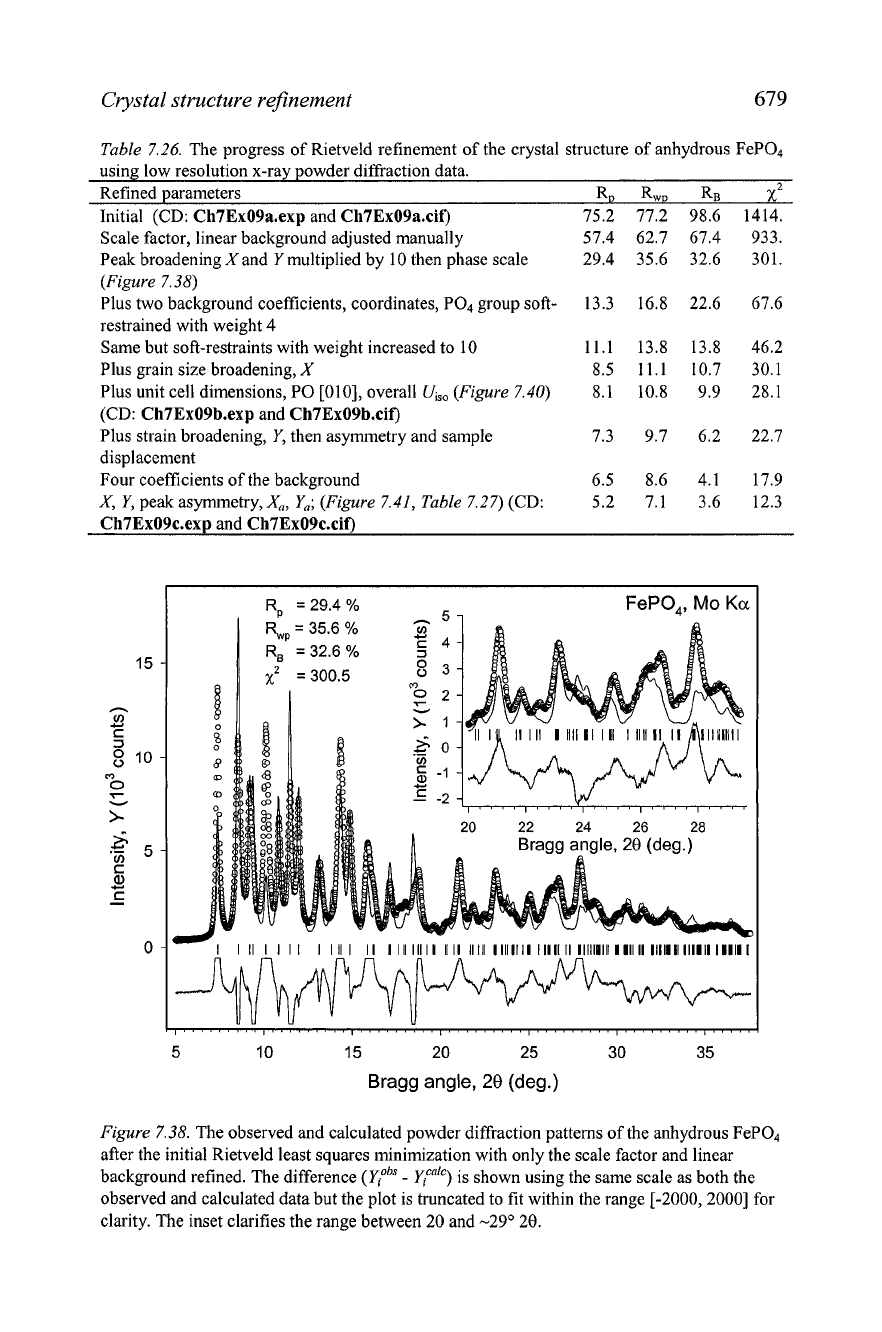
Figure 7.38.
The observed and calculated powder diffraction patterns of the anhydrous FeP04
after the initial Rietveld least squares minimization with only the scale factor and linear
background refined. The difference
(yiobS
-
&"lc)
is shown using the same scale as both the
observed and calculated data but the plot is truncated to fit within the range
[-2000,2000]
for
clarity. The inset clarifies the range between
20
and
-29" 20.

680
Chapter
7
Both the complexity and low resolution of the experimental data, coupled
with the possibility of far fi-om ideal coordinates of some or all atoms in the
optimized model of the crystal structure' present an interesting dilemma in
the selection of the next set of parameters for a subsequent Rietveld
refinement. Although it is obvious that profile parameters require further
improvement, it also appears that both the inadequacy of the initial fit and
low resolution of the data may not allow their unequivocal refinement. On
the other hand, atomic coordinates likely deviate significantly from their real
values, which is easily seen in
Figure
7.38
indicating significant
discrepancies between the observed and calculated intensities for many
Bragg reflections.
Hence, the following refinement step included a linear background and
coordinates of all atoms.2 In anticipation of considerable problems with the
least squares minimization and high probability of moving away from a
global minimum, soft restraints were employed to restrain the well-known
geometry of the phosphate group
P04.3 Its initial geometry, obtained as a
result of quantum chemical optimization, was nearly perfect: the
P-0
distances vary between 1.52 and 1.54 A, while the 0-P-0 angles were
between 107.8 and 110.2". The following restrains were imposed: the
P-0
distance of 1.53
h
0.01 A, and the 0-P-0 angles of 109.5
*
2.0'; the weight
was set to
4.
The first five cycles of the refinement substantially improve the
fit, lowering R, by more than 20
%,
down to 16.8
%.
This reduction,
however, comes at the cost of worsening the PO4 geometry: the P-0
distances now range from 1.43 to 1.61
A
and the 0-P-0 angles vary from
103 to 117". The Fe-0 distances remain acceptable, and they range from
1.83 to 1.95
A
but one additional elongated Fe-0 bond of 2.27
A
emerges.
In
order to improve the geometry, the soft restraint weight factor was
increased to 10, and several subsequent least squares cycles were conducted.
The weighted residual further decreases and, most importantly, the geometry
of the
PO4 group recovers. The correctness of this adjustment is
demonstrated in
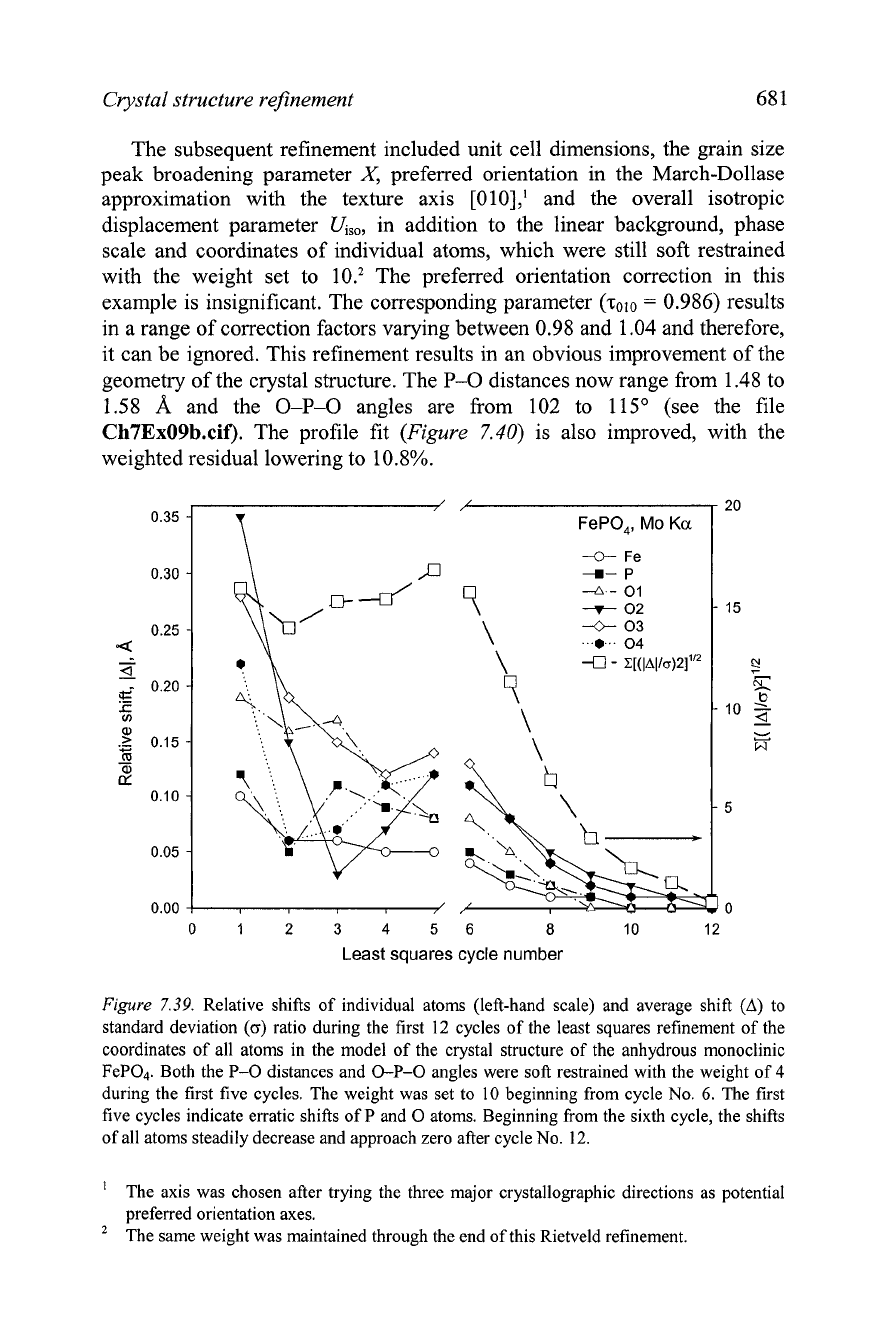
Figure
7.39, which illustrates relative shifts of all atoms as
functions of least squares cycle number. It is obvious, that setting the weight
to 4 does little
to
stabilize the convergence, while increasing the weight to 10
results in a rapid reduction of the magnitudes of atomic displacements over a
few refinement cycles.
'
It is worthy of reminding one that the quantum chemical optimization of the geometry has
been performed after Fe was substituted by
Al.
Positions (coordinates) of atoms in the unit cell are the strongest contributors into the
computed integrated intensities of Bragg reflections assuming that preferred orientation
effects are weak. For this powder, preferred orientation was expected (and later found) to
be minor due to small particle sizes and predominantly isotropic particle shapes.
A
thorough reader should be able to verify the correctness of this statement by attempting
Rietveld refinement without imposing soft restraints.
Crystal structure refinement 68 1
The subsequent refinement included unit cell dimensions, the grain size
peak broadening parameter
X,
preferred orientation in the March-Dollase
approximation with the texture axis [OlO],' and the overall isotropic
displacement parameter
Ui:,,,,
in addition to the linear background, phase
scale and coordinates of individual atoms, which were still soft restrained
with the weight set to
10.' The preferred orientation correction in this
example is insignificant. The corresponding parameter (~0~0
=
0.986) results
in a range of correction factors varying between 0.98 and 1.04 and therefore,
it can be ignored. This refinement results in an obvious improvement of the
geometry of the crystal structure. The
P-0
distances now range from 1.48 to
1.58
A
and the
0-P-0
angles are from 102 to 115' (see the file
Ch7ExO9b.cif). The profile fit (Figure
7.40)
is also improved, with the
weighted residual lowering to 10.8%;
0123456
8
10 12
Least squares cycle
number
Figure
7.39.
Relative shifts of individual atoms (left-hand scale) and average shift
(A)
to
standard deviation
(o)
ratio during the first
12
cycles of the least squares refinement of the
coordinates of all atoms in the model of the crystal structure of the anhydrous monoclinic
FeP04. Both the P-0 distances and 0-P-0 angles were soft restrained with the weight of
4
during the first five cycles. The weight was set to
10
beginning from cycle No.
6.
The first
five cycles indicate erratic shifts of P and
0
atoms. Beginning from the sixth cycle, the shifts
of all atoms steadily decrease and approach zero after cycle No.
12.
'
The axis was chosen after trying the three major crystallographic directions as potential
preferred orientation axes.
'
The same weight was maintained through the end of this Rietveld refinement.