Pecharsky V.K., Zavalij P.Y. Fundamentals of Powder Diffraction and Structural Characterization of Materials
Подождите немного. Документ загружается.


662
Chapter
7
7.9
Rietveld refinement and completion of the
rn~~Mo,O~~
structure1
In
this section, we will illustrate the Rietveld refinement of a complex
molybdenum oxide-based layered structure containing 15 independent Mo
and
0
atoms with an intercalated organic methyl ammonium ion (one N, one
C and 6 H atoms) using conventional x-ray powder diffraction data. The
model of the Mo7OZ2 framework was obtained from the assumption that it is
nearly identical to that found in the T12M07022 compound, as identified by a
search of the ICSD database (section 6.15). Structure determination should
be completed by locating the methyl ammonium (CH3NH3f) ion from a
difference Fourier map and refining all relevant parameters. When located in
the unit cell, the N atom is clearly distinguished from carbon by much
shorter distances the former makes with oxygen, because of
N-H...O
hydrogen bonding.
The experimental powder diffraction pattern was collected from
7
to 100'
28 with a 0.02' step and a counting time varying from 30 seclstep at low
Bragg angles to 60 seclstep at high angles, scaled after the data collection to
a constant 30 seclstep counting time. The following parameters were used at
the beginning of this refinement:
The initial model of the structure (the molybdenum oxide layer) was
taken from
Table
6.42.
The default profile parameters were taken from the instrumental
parameter file Scintag.prm as described in section
7.6.
The sample shift parameter
S,
=
4.49
for the sample displacement s
=
-0.098
mm, was obtained together with the unit cell dimensions during
lattice parameters refinement.
The space group is
C21c and the unit cell dimensions are
a
=
23.065,
b
=
5.5134,
c
=
19.561
A,
/3
=
122.93" as determined earlier.
The overall isotropic atomic displacement parameter was assumed at
Uiso=
0.015 A'.
The starting model with all necessary parameters is found on the CD in
the files Ch7Ex07a.e~~ and Ch7Ex07a.cif, and the experimental pattern is
in the file Ch7ExO7-CuKa.raw. Initially, only the phase scale factor and 6
coefficients of the shifted-Chebyshev polynomial to approximate the
background were refined, resulting in relatively high residuals, which are
listed in
Table
7.22.
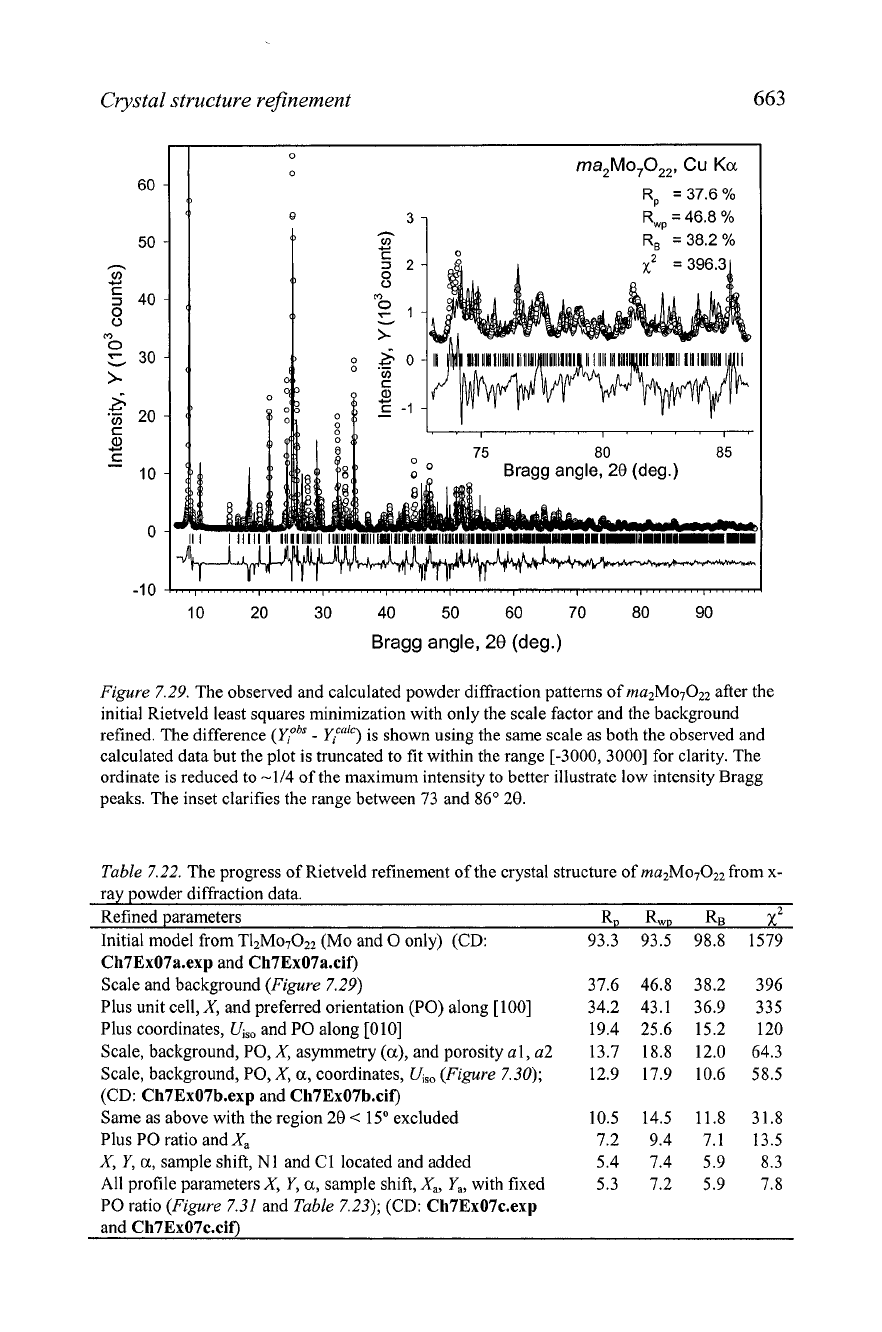
The poor fit is mainly due to a mismatch between the
observed and calculated intensities as can be seen in
Figure
7.29.
'
P.Y.
Zavalij and MS. Whittingham, The crystal structure of layered methylammonium
molybdate (CH3NH3)2M07022 from X-ray powder data, Acta Cryst.,
C53,
1374
(1997)
Crystal structure refinement
663
ma,Mo,O,,,
Cu
Ka
R,
=
37.6
%
R,,
=
46.8
%
Bragg angle,
20
(deg.)
II I
I
11
11
1
II
111
11 11111111 llllllllllllllll
lUlllllllllllUlllllllUillllllwMlII~IIIIB11
I
I""""'I'""""I""""'I"~""","'""'~,~~~~~l~~~"~"~I~"~
,,,,',.','''''
10 20 30 40 50 60 70 80 90
Bragg angle,
28
(deg.)
Figure 7.29. The observed and calculated powder diffraction patterns of ma2M07022 after the
initial Rietveld least squares minimization with only the scale factor and the background
refined. The difference
(YiobS
-
&ca'c)
is shown using the same scale as both the observed and
calculated data but the plot is truncated to fit within the range [-3000, 30001 for clarity. The
ordinate is reduced to -114 of the maximum intensity to better illustrate low intensity Bragg
peaks. The inset clarifies the range between 73 and 86' 20.
Table 7.22. The progress of Rietveld refinement of the crystal structure of
from
x-
ray powder diffraction data.
Refined parameters RD RWD
R~
x2
Initial model from T12M07022 (Mo and
0
only) (CD: 93.3 93.5 98.8 1579
Ch7Ex07a.e~~
and
Ch7Ex07a.cif)
Scale and background (Figure 7.29) 37.6 46.8 38.2 396
Plus unit cell,
X,
and preferred orientation (PO) along [I001 34.2 43.1 36.9 335
Plus coordinates,
Ui,,
and PO along [010] 19.4 25.6 15.2 120
Scale, background, PO,
X,
asymmetry (a), and porosity a1, a2 13.7 18.8 12.0 64.3
Scale, background, PO,
X,
a,
coordinates,
Ui,,
(Figure 7.30); 12.9 17.9 10.6 58.5
(CD:
Ch7Ex07b.e~~
and
Ch7Ex07b.cif)
Same as above with the region 20
<
15" excluded 10.5 14.5 11.8 31.8
Plus PO ratio and
X,
7.2 9.4 7.1 13.5
X,
Y,
a,
sample shift,
N1
and C1 located and added 5.4 7.4 5.9 8.3
All profile parameters
X,
Y,
a,
sample shift,
X,,
Y,,
with fixed 5.3 7.2 5.9 7.8
PO ratio (Figure 7.31 and Table 7.23);
(CD:
Ch7Ex07c.e~~
and
Ch7Ex07c.cif)

664 Chapter
7
It is highly likely that the intensity mismatches are caused by a relatively
crude initial structural model since the atomic coordinates were taken
directly from the T1-based structure, even without correcting for the lattice
distortion along
a
(compare the unit cell dimensions of both materials in
Table 6.41). Furthermore, a considerable preferred orientation could be
expected because of yet another distinctly layered structure (see Figure
6.30). Therefore, the subsequent refinement included unit cell dimensions,
grain size contribution to peak broadening
(3
and preferred orientation (PO)
parallel to the [loo] direction, along which the M07022 layers are stacked.
Some improvement of the fit has been observed as a result. Unlike in the two
examples considered in sections 7.7 and 7.8, the preferred orientation is not
as strong here, possibly due to a less severe cleaving of the particles during
grinding.
A noticeable improvement, especially in the Bragg residual, occurs when
the coordinates of all atoms have been refined together with the isotropic
displacement parameters: individual for the Mo atoms and one parameter,
common for all oxygen atoms. A second preferred orientation parameter
along the
[OlO] axis was added as well, but its effect on the improvement of
the fit was quite small.
A
slightly negative overall isotropic displacement
parameter, U,,,
=
-0.002(4) A2, for the
0
atoms likely indicates a
contribution from the specimen porosity, which to a certain extent, also
incorporates other unaccounted systematic errors, e.g. absorption and beam
size exceeding the sample dimensions at low Bragg angles due to an
improper selection of the divergence slit aperture. Therefore, the porosity
effect was optimized in a subsequent refinement using two parameters
(a,
and a2) in the Suortti approximation. The latter refinement was carried out
after isotropic atomic displacement parameters were set to 0.015
A2
for all
molybdenum and 0.020
A2
for all oxygen atoms, and all atomic parameters
were kept fixed. The two porosity coefficients were refined to al
=
0.3 1 and
a2
=
0.16 starting from the initial 0.40 and 0.40, respectively, after which
they were kept fixed through the end of the Rietveld refinement. Next, the
individual atomic parameters were released and re-refined. This substantially
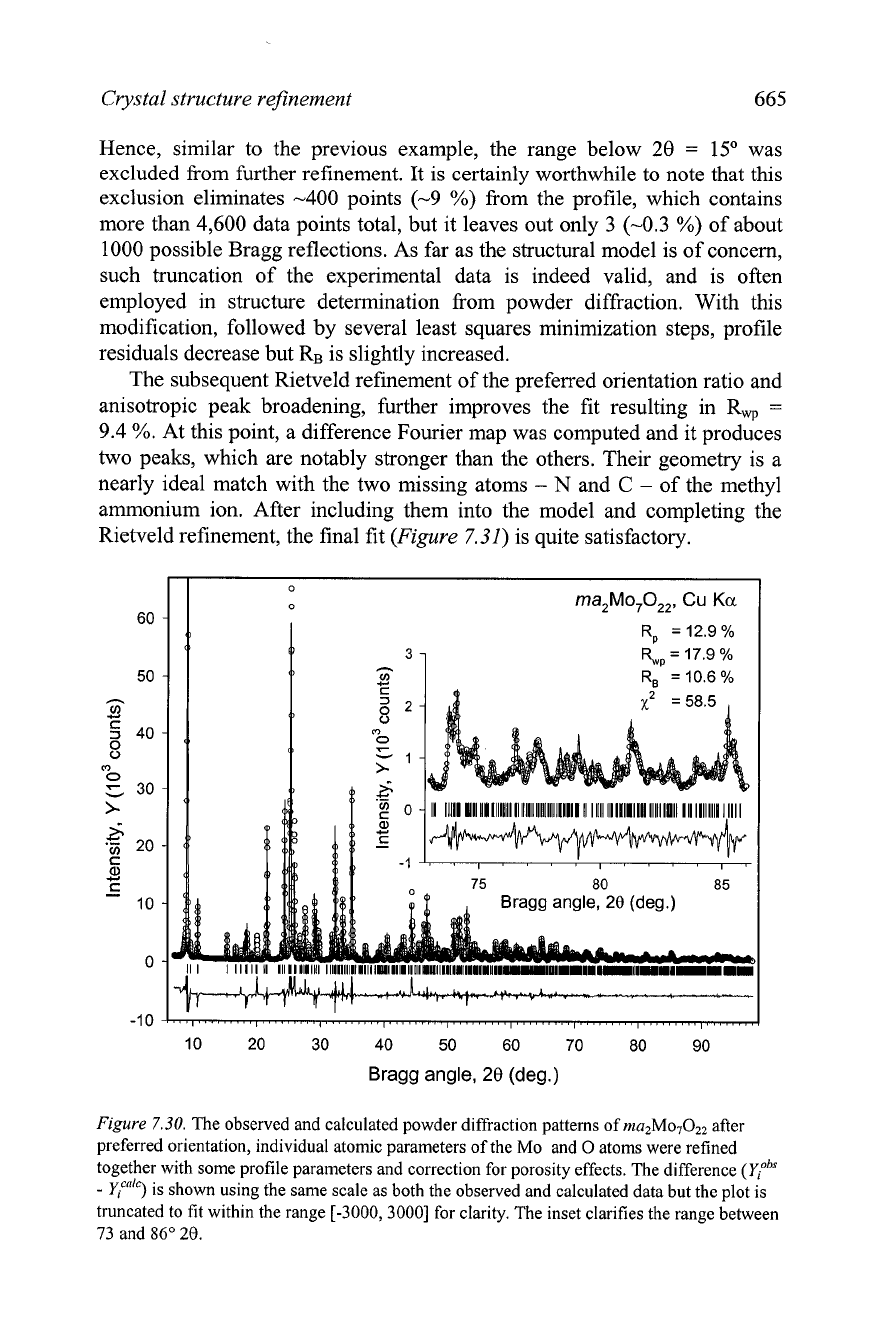
improves the fit as shown in Figure 7.30.
Regardless of the considerable improvement, some mismatches between
the observed and computed intensities remain. At this point, we are still
missing one nitrogen and one carbon from the methyl ammonium ion, not
counting six hydrogen atoms, which will be nearly impossible to locate from
x-ray powder data (see section 7.7). Pinpointing the locations of carbon and
nitrogen in the unit cell and their inclusion into the model should indeed
improve the fit. However, another potentially deleterious effect on the
overall fit is the presence of a large intensity peak at low Bragg angle (28
E
go),
in front of which there is a much smaller and broader impurity peak.
Crystal structure refinement
665
Hence, similar to the previous example, the range below
20
=
15" was
excluded from further refinement. It is certainly worthwhile to note that this
exclusion eliminates -400 points (-9
%)
from the profile, which contains
more than 4,600 data points total, but it leaves out only 3 (-0.3
%)
of about
1000 possible Bragg reflections. As far as the structural model is of concern,
such truncation of the experimental data is indeed valid, and is often
employed in structure determination from powder diffraction. With this
modification, followed by several least squares minimization steps, profile
residuals decrease but
RB
is slightly increased.
The subsequent Rietveld refinement of the preferred orientation ratio and
anisotropic peak broadening, further improves the fit resulting in
R,
=
9.4
%.
At this point, a difference Fourier map was computed and it produces
two peaks, which are notably stronger than the others. Their geometry is a
nearly ideal match with the two missing atoms
-
N
and C
-
of the methyl
ammonium ion. After including them into the model and completing the
-
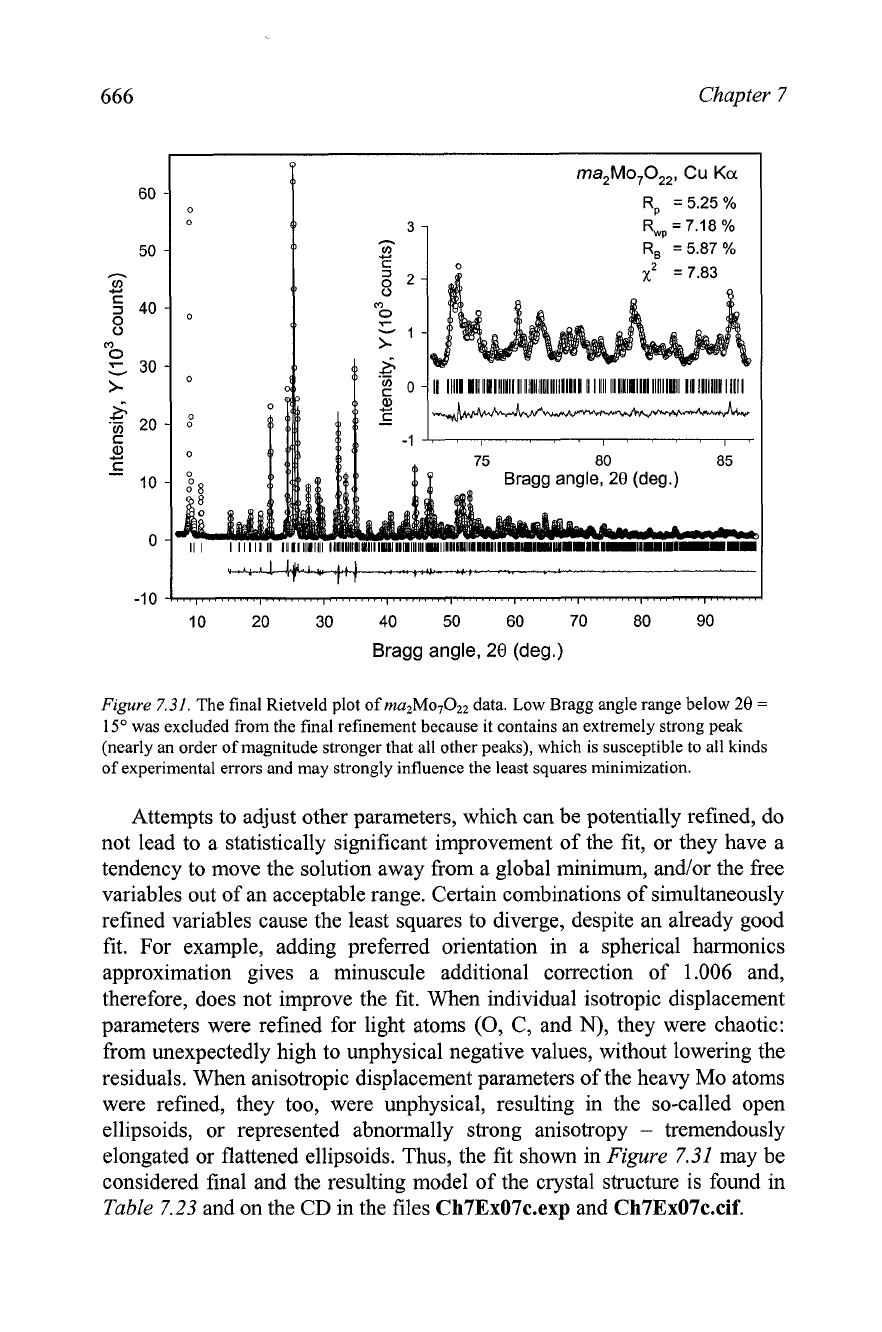
Rietveld refinement, the final fi<(~i~ure
7.31)
is quite satisfactory-.
ma2Mo,02,,
Cu
Ka
R,
=12.9%
&,=
17.9%
R,
=
10.6
%
Bragg angle,
28
(deg.)
Figure
7.30.
The observed and calculated powder diffraction patterns of rn~~Mo~0,~ after
preferred orientation, individual atomic parameters of the Mo
and
0
atoms were refined
together with some profile parameters aid correction for porosity effects. The difference
(yrbS
-
Y,~"")
is shown using the same scale as both the observed and calculated data but the plot is
truncated to fit within the range [-3000,3000] for clarity. The inset clarifies the range between
73 and
86"
28.
Chapter
7
Bragg angle,
20
(deg.)
I1 I
1
I I
I
I
I
II 111 11 1111111
llllllllllu~11111llllUllllllllllUllllllll1mwllllUmlII
I""""'I""""'I"""'"I'""""I""""'I'"~"~"I""""',""""'l"""'
10 20 30 40 50 60 70 80 90
Bragg angle,
28
(deg.)
Figure
7.31.
The final Rietveld plot of
data. Low Bragg angle range below
20
=
15"
was excluded from the final refinement because it contains an extremely strong peak
(nearly an order of magnitude stronger that all other peaks), which is susceptible to all kinds
of experimental errors and may strongly influence the least squares minimization.
Attempts to adjust other parameters, which can be potentially refined, do
not lead to a statistically significant improvement of the fit, or they have a
tendency to move the solution away from a global minimum,
andlor the free
variables out of an acceptable range. Certain combinations of simultaneously
refined variables cause the least squares to diverge, despite an already good
fit. For example, adding preferred orientation in a spherical harmonics
approximation gives a minuscule additional correction of 1.006 and,
therefore, does not improve the fit. When individual isotropic displacement
parameters were refined for light atoms
(0,
C, and
N),
they were chaotic:
from unexpectedly high to unphysical negative values, without lowering the
residuals. When anisotropic displacement parameters of the heavy Mo atoms
were refined, they too, were unphysical, resulting in the so-called open
ellipsoids, or represented abnormally strong anisotropy
-
tremendously
elongated or flattened ellipsoids. Thus, the fit shown in
Figure
7.31
may be
considered final and the resulting model of the crystal structure is found in
Table
7.23
and on the CD in the files
Ch7Ex07c.e~~
and
Ch7Ex07c.cif.
Crystal structure refinement
667
Table
7.23.
Final atomic parameters and interatomic distances (in A) of ma2M07022 obtained
from Rietveld refinement using GSAS. The refined unit cell parameters are: a
=
23.0707(3),
b
=
5.51522(7),
c
=
19.5669(2)
A,
=
122.930(1)", V
=
2089.68(5)
A3,
space group C21c. All
crystallographic data can be also found on the
CD
in the files
Ch7Ex07c.e~~
and
Ch7Ex07c.cif.
Atom
x
Y
z
U~SO
Bond distances,
A
Mol Mo2 Mo3 Mo4
clb 0.265(1) 0.143(4) 0.196(2) 0.014(4)
a
Hydrogen bond distances for
N1
are: 2.97(3)
A
to 05, 2.88(3)
A
to 02, 2.85(3)
A
to 010,
and 2.76(2)
A
to 01 1.
C1 distances: 1.46(3)
A
to N1,3.31(4)
A
to 02,3.25(3) A to 01, and 3.39(3) A to 02.
The correctness of the crystal structure is doubtless because the M070~~
layer is isotypical to both T1- and Cs-based compounds (see Table
6.41).
The
nearly identical layer also exists in the intercalate, containing a larger
organic molecule:
RMO~O~~.H~O, where
R
=
4,4'-bipyridinium (H-NC5H4-
C~H~N-H)".' The orientation of the methyl ammonium molecule (in other
words, the recognition of the nitrogen and carbon atoms) is also convincing
because the N atom is much closer to the
0
atoms than the
C
atom, which is
due to the formation of strong N-H.a.0 hydrogen bonds. The C-H...O
interactions are much weaker, as follows from the comparison of bond
lengths: 2.8 to 2.9
A
for N--.O and 3.3
A
or more for the C...O distances.
The
C-N
bond length, 1.46(3) A, is within the expected range (compare to
single crystal data for maV3O7, where the C-N bond length is 1.487(8)
A).
Stacking of the layers with the intercalated methyl ammonium ions forming
'
P.J.
Zapf,
R.C.
Haushalter, and
J.
Zubieta. Crystal engineering of inorganiclorganic
composite solids: the structure-directing role of aromatic ammonium cations in the
synthesis of the "stepv-layered molybdenum oxide phase
[4,4'-H2bpy][Mo7022].H20,
Chem. Commun., 321 (1997)

668 Chapter
7
N-H...O
hydrogen bonds, which hold the layers together, is illustrated in
Figure
7.32.
The peculiarity of this refinement is the relatively large unit cell with 17
non-hydrogen atoms in the asymmetric unit, resulting in 1035 possible
independent Bragg reflections and a total of 75 free least squares variables,
respectively. This example also illustrates that the location and the quality of
the determination of light
C
and
N
atoms, forming a small organic molecule
encapsulated between massive molybdenum oxide layers, is quite reliable,
provided sufficient quality powder diffraction data are available. The
preferred orientation in this case is less severe when compared to the two
previous examples:
zloo
=
0.690(3) and
zolo
=
1.091(4) with an approximate
1 :2 ratio, which corresponds to the correction range between 0.7 to 1.8, or to
the preferred orientation magnitude of about 2.6. The latter value is still
significant and it may be partially responsible for the unphysical anisotropic
displacement parameters of the Mo atoms.
Figure
7.32.
The crystal structure of
shown along the Y-axis in perspective view.
The Mo atoms are depicted as large dark spheres located inside translucent octahedra formed
by oxygen atoms (small light spheres). The
N
atoms are shown as small black spheres and
the
C
atoms as small grey spheres. The
N-[H]...O
hydrogen bonds are shown using thin lines
without hydrogen atoms. Unlike in the crystal structure of the
tmaV307
(see section
7.8),
the
coordinates of hydrogen atoms cannot be easily computed because methylammonium ion can
rotate around the
C-N
bond, although this rotation is restrained by the formation of
N-H...O
hydrogen bonds. Hydrogen atoms could not be located from a differential Fourier map
because of the presence of the large amount of strongly scattering atoms (Mo)
in
the material.

Crystal structure repnernent 669
7.10
Rietveld refinement
of
Mn7(OH)3(V04)41
Using this example and employing conventional x-ray powder data, we
will illustrate Rietveld refinement of a medium complexity inorganic
structure with 10 independent atoms occupying various sites in an acentric
hexagonal space group symmetry. Several sites are occupied partially and
therefore, we will learn how to perform a sensible refinement of the
chemical composition. The refinement begins from the isostructural and
fully ordered zinc-vanadate-sulfate crystal structure, as was established in
the previous chapter (section 6.16). During the least squares minimization,
partially vacant sites were identified and their populations were refined in
the first approximation independently, and then with reasonable restrictions
on chemical composition, until the complete convergence was achieved. The
correctness of the crystal structure determination from powder data was later
confirmed by single crystal diffraction work. This example also shows how
some of the constraints, realized in
GSAS,
can be invoked.
The idealized chemical composition of the title compound when all sites
are fully occupied is Mn7(OH)3(V04)4. The experimental data were collected
with a 0.01" step.
In
the range from 12 to 70"
28
the counting time was 15
seclstep, and it was increased to 30 seclstep from 70 to 132' 28.
In
this way,
it was possible to achieve better counting statistics at high Bragg angles,
where the scattered intensity is significantly reduced, while simultaneously
avoiding a potential for errors associated with, for example, continuously
varying or different divergence slit apertures. The high Bragg angle range
was scaled to a fixed counting time of
15
seclstep for all computations. The
following parameters were used at the beginning of this refinement:
-
The initial structural model was taken from Zn7(OH)3(S04)(V04)3, which
crystallizes in the same (P63mc) space group with
a
=
12.813 and
c
=
5.1425 8L, by replacing Zn with Mn and S with
V,
as was assumed in the
previous chapter (Table 6.44).
-
The default profile parameters were taken from the instrumental
parameter data file
Scintag-prm,
as described above in section 7.6.
-
The sample shift parameter S,
=
5.13 for the sample displacement s
=
-0.1 12 mm, which was obtained together with the unit cell dimensions at
an earlier stage of the structure solution.
-
The space group is P63mc and the unit cell dimensions are a
=
13.229
and c
=
5.2553 8L, also determined earlier.
-
The overall isotropic displacement parameter
U,,,
=
0.0 15 8L2.
'
F.
Zhang,
P.Y.
Zavalij, MS. Whittingham. Synthesis and characterization of a pipe-
structure manganese vanadium oxide by hydrothermal reaction.
J.
Mater. Chem.,
9,
3
137
(1999).
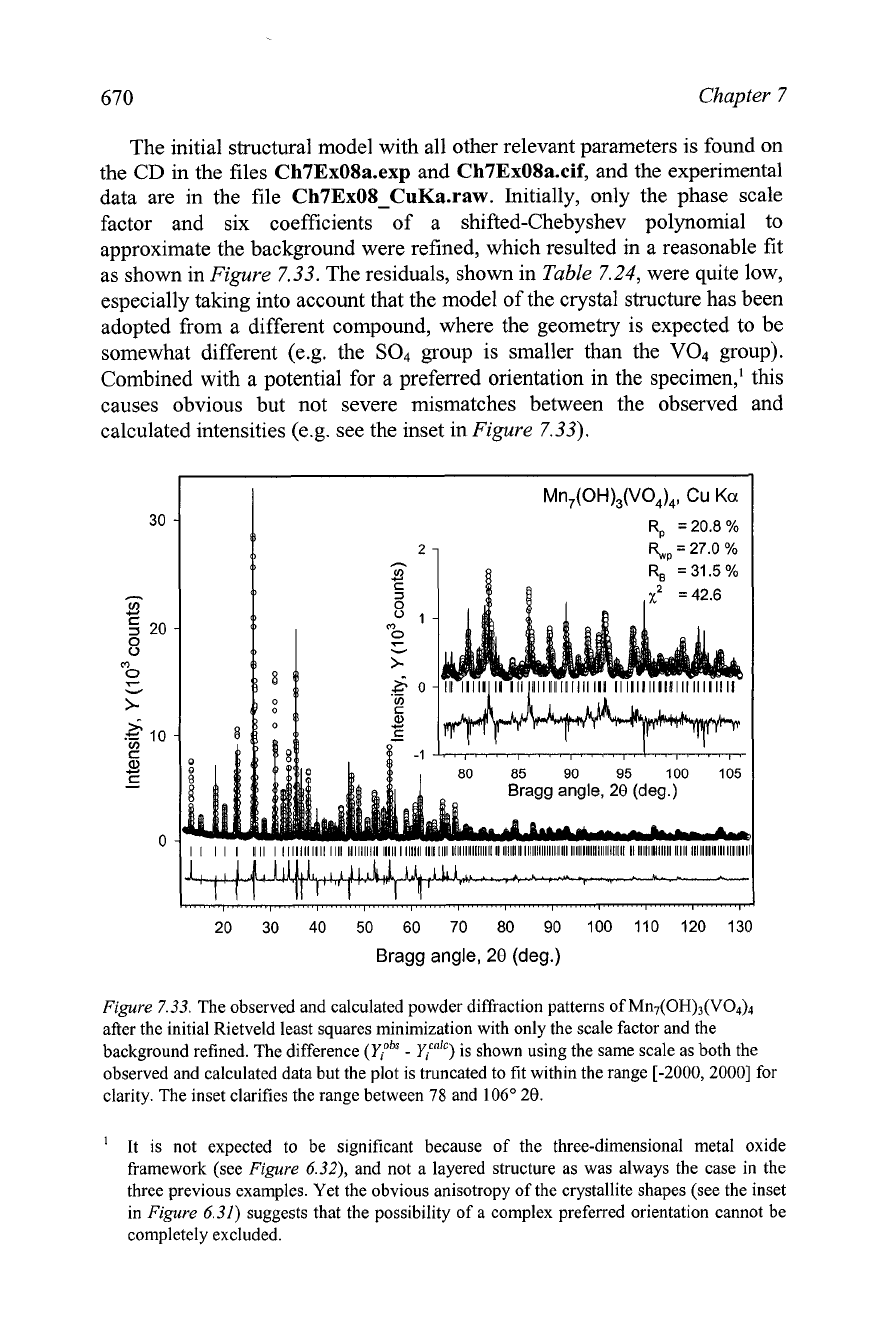
670
Chapter
7
The initial structural model with all other relevant parameters is found on
the
CD
in the files Ch7ExOSa.exp and Ch7ExOSa.cif, and the experimental
data are in the file Ch7Ex08-CuKa.raw. Initially, only the phase scale
factor and six coefficients of a shifted-Chebyshev polynomial to
approximate the background were refined, which resulted in a reasonable fit
as shown in
Figure
7.33. The residuals, shown in
Table
7.24,
were quite low,
especially taking into account that the model of the crystal structure has been
adopted from a different compound, where the geometry is expected to be
somewhat different
(e.g. the
SO4
group is smaller than the
V04
group).
Combined with a potential for a preferred orientation in the specimen,' this
causes obvious but not severe mismatches between the observed and
calculated intensities (e.g. see the inset in
Figure
7.33).
5
1-
m
s
-
>
2;
0-
.-
(I)
C
a,
C
t
-
80 85 90 95 100 105
Bragg angle,
20
(deg.)
I
I
I I
II
U
II
1
I IIIIII
1111
1
Ill
1111111111
11111
I11l111
1111
1111
1111111111111111
11
1111111111111111111lllllll
1111111111111111111lll II IIIIIIIIIIIII
11111
1111111111111111111
,
1.
I
,I
,I
I
20 30 40 50 60 70 80 SO 100 110 120 130
Bragg angle,
28
(deg.)
Figure
7.33. The observed and calculated powder diffraction patterns of M~I~(OH)~(VO&
after the initial Rietveld least squares minimization with only the scale factor and the
background refined. The difference (Y:~'
-
Y?) is shown using the same scale as both the
observed and calculated data but the plot is truncated to fit within the range
[-2000,2000]
for
clarity. The inset clarifies the range between
78
and
106"
28.
'
It is not expected to be significant because of the three-dimensional metal oxide
framework (see
Figure
6.32), and not a layered structure as was always the case in the
three previous examples. Yet the obvious anisotropy of the crystallite shapes (see the inset
in
Figure
6.31) suggests that the possibility of a complex preferred orientation cannot be
completely excluded.
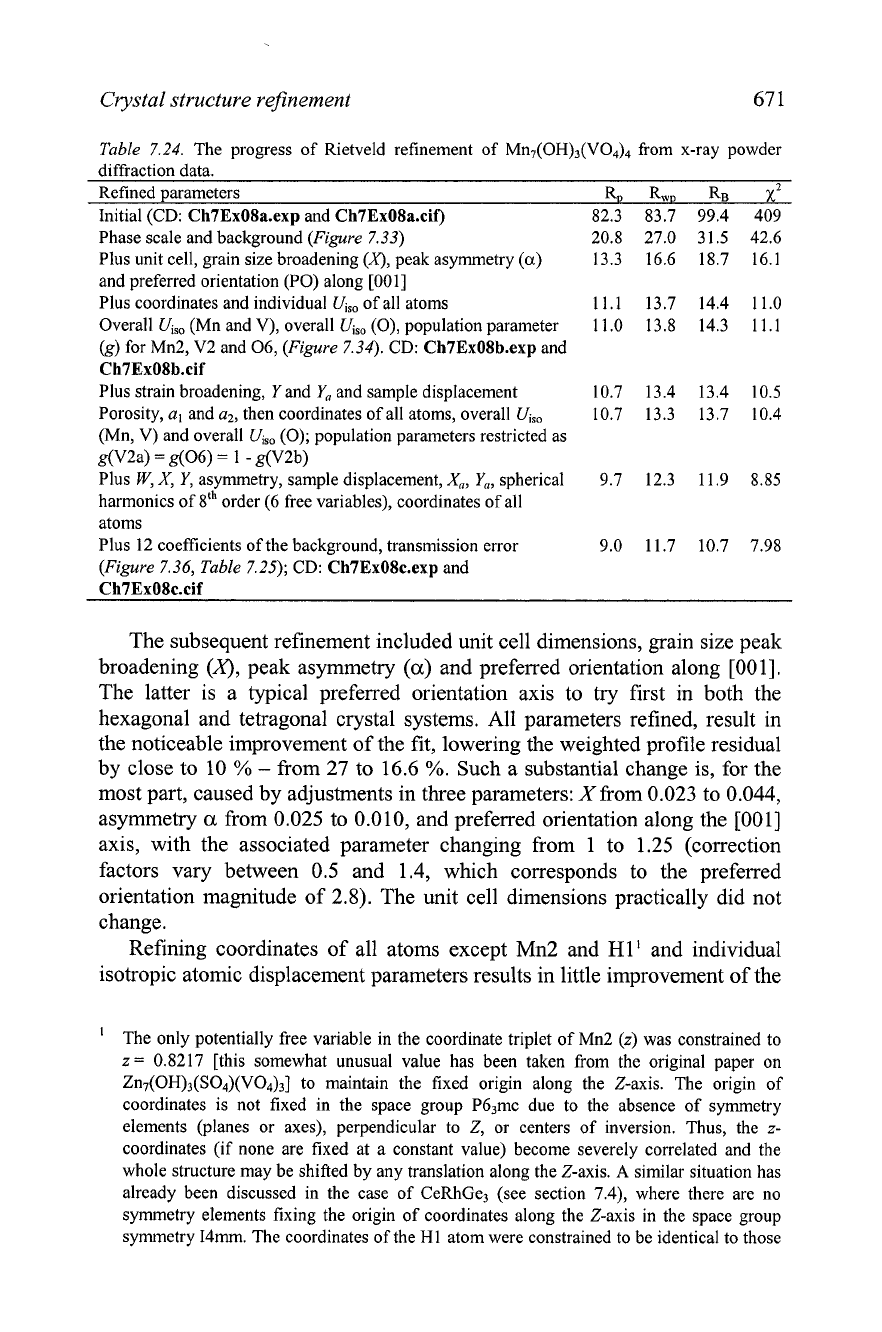
Crystal structure refinement
67
1
Table 7.24. The progress of Rietveld refinement of Mn7(0H)3(V04)4 from x-ray powder
diffraction data.
Refined parameters
%
RwD RB
xZ
Initial (CD:
Ch7ExOSa.exp
and
Ch7ExOSa.cif)
82.3 83.7 99.4 409
Phase scale and background (Figure 7.33) 20.8 27.0 31.5 42.6
Plus unit cell, grain size broadening
(3,
peak asymmetry
(a)
13.3 16.6 18.7 16.1
and preferred orientation (PO) along [OOl]
Plus coordinates and individual
Uiso
of all atoms 11.1 13.7 14.4 11.0
Overall
Uis0
(Mn and V), overall
Uiso
(0), population parameter 1 1.0 13.8 14.3 1 1.1
(g) for Mn2, V2 and 06, (Figure 7.34). CD:
Ch7ExOSb.exp
and
Ch7ExOSb.cif
Plus strain broadening,
Y
and
Y,,
and sample displacement 10.7 13.4 13.4 10.5
Porosity, a, and a2, then coordinates of all atoms, overall
Uiso
10.7 13.3 13.7 10.4
(Mn, V) and overall
Uis0
(0); population parameters restricted as
g(V2a)
=
g(06)
=
1
-
g(V2b)
Plus
W,
X,
Y,
asymmetry, sample displacement,
X,,
Y,,
spherical 9.7 12.3 1 1.9 8.85
harmonics of 8th order (6 free variables), coordinates of all
atoms
Plus 12 coefficients of the background, transmission error 9.0 11.7 10.7 7.98
(Figure 7.36, Table 7.25);
CD:
Ch7ExOSc.exp
and
Ch7ExOSc.cif
The subsequent refinement included unit cell dimensions, grain size peak
broadening
(3,
peak asymmetry
(a)
and preferred orientation along [OOl].
The latter is a typical preferred orientation axis to try first in both the
hexagonal and tetragonal crystal systems. All parameters refined, result in
the noticeable improvement of the fit, lowering the weighted profile residual
by close to 10
%
-
from
27
to 16.6
%.
Such a substantial change is, for the
most part, caused by adjustments in three parameters: Xfrom 0.023 to 0.044,
asymmetry
a
from 0.025 to 0.010, and preferred orientation along the [OOl]
axis, with the associated parameter changing from 1 to 1.25 (correction
factors vary between 0.5 and 1.4, which corresponds to the preferred
orientation magnitude of 2.8). The unit cell dimensions practically did not
change.
Refining coordinates of all atoms except Mn2 and
H1'
and individual
isotropic atomic displacement parameters results in little improvement of the
'
The only potentially free variable in the coordinate triplet of Mn2
(z)
was constrained to
z
=
0.8217 [this somewhat unusual value has been taken from the original paper on
Zn7(OH)3(S04)(V04)3] to maintain the fixed origin along the Z-axis. The origin of
coordinates is not fixed in the space group P63mc due to the absence of symmetry
elements (planes or axes), perpendicular to Z, or centers of inversion. Thus, the
z-
coordinates (if none are fixed at a constant value) become severely correlated and the
whole structure may be shifted by any translation along the Z-axis.
A
similar situation has
already been discussed in the case of CeRhGe3 (see section 7.4), where there are no
symmetry elements fixing the origin of coordinates along the Z-axis in the space group
symmetry I4mm. The coordinates of the HI atom were constrained to be identical to those