Pecharsky V.K., Zavalij P.Y. Fundamentals of Powder Diffraction and Structural Characterization of Materials
Подождите немного. Документ загружается.


642 Chapter
7
Both damping techniques stabilize Rietveld refinement but obviously
require more least squares cycles for its completion.
It
is worth noting, that
according to the GSAS default (which can be changed), the refinement
process is considered converged to a global minimum when the maximum
shift
(Axi) observed among all free variables is less than 11100~~ of the
corresponding standard deviation (oxi), i.e. when IAxilloxi
<
0.01, i
=
1, 2,
. .
.
p
andp is the number of free variables.'
Structure and properties of both the metal oxides and their intercalates,
which are used as examples in sections 7.7 to 7.1 1, result in high anisotropy
of crystal shapes. Distinct, plate-like or needle-like shapes of particles
(Figure 6.23, Figure 6.26, and Figure 6.31) cause a highly non-random
distribution of particle orientations even after thorough grinding and
screening. The state of the specimen often cannot be adequately described
using a simple single-parameter preferred orientation
model(s) and two
preferred orientation axes or a spherical harmonics expansion should be
employed.
In
parallel, the anisotropy of the particle shapes almost
necessarily causes the anisotropy in the broadening of the diffraction peaks,
which, in order to obtain a good fit, has to be accounted for as well.
Unless noted to the contrary, the initial values of peak shape parameters
used in the following five examples, were obtained from a refinement using
the experimental powder diffraction pattern of LaB6 measured on a Scintag
XDS2000 diffractometer employing Cu
Ka
radiation and a liquid nitrogen-
cooled Ge(Li) solid-state detector at typical experimental settings. These are
found on the
CD
in the file
Scintag.prm.
We note that all profile parameters
employed in GSAS are in centidegrees (i.e. the corresponding values in
degrees have been multiplied by 100). Thus, the following settings and
initial parameters were used:
-
The peak shape function was a Thompson modified pseudo-Voigt.' It is
referred as No. 2 in GSAS (also see section 2.9.1 and relevant equations).
-
Bragg peaks were extended in the range where their calculated intensity
was greater or equal to 0.5% of the intensity at the peak maximum.
-
The instrumental component of
FWHM
was given by
U
=
0.7104,
V
=
-0.9565,
W=
1.7318;
P=
0.
-
Isotropic peak broadening due to grain size and strain with
X=
2.2952,
Y
=
3.955 1, and
X,
=
0 and
Y,
=
0.
-
Peak asymmetry
a
=
2.5471.
-
Porosity and absorption effects were initially accounted for by using the
Suortti approach (see Eq. 2.76) with parameters a,
=
0.4 and a:!
=
0.4.
IUCr imposes the requirement
Ihxilloxi
<
0.05
to ensure completeness of refinements
based on single crystal data. In Rietveld
fits,
(Axi(loxi
<
0.1
are quite satisfactory.
P.
Thompson,
D.E.
Cox, and
J.B.
Hastings. Rietveld refinement of Debye-Scherrer
synchrotron x-ray data from A1203,
J.
Appl. Cryst.
20,79
(1987).

Crystal structure rejnement
643
These two parameters have a tendency to strong correlation, and they
were refined only when the quality of the pattern was sufficiently high.
The sample displacement parameter S, calculated for each particular case
from the displacement (s, in mm) obtained during unit cell refinement as:
S,
=
-36000s/(nR) or S,
=
-144sln for the goniometer radius
R
=
250
mrn.
Cu Ka radiation wavelengths used were 1 S40562 and 1 S4439O
A
for
Kal
and Ka2 components, respectively.
The initial value of the phase scale factor was always 1
.I
The initial background was set to
a
constant value of 100 counts or, in
some difficult cases, fitted manually and kept fixed during initial
refinement steps.
Unless noted to the contrary, a 6-parameter shifted-Chebyshev function
was employed to fit the background at later refinement stages.
The initial atomic displacement parameters were always set to
U,,,
=
0.015 A2, which is equivalent to Biso
(=
8n2Uiso) of -1.2
A2.
Fractional population factors in GSAS are treated as g's (see Eq. 7.8),
while site multiplicities are automatically accounted for and cannot be
changed. Site populations, however, can be refined when needed. For
example, a population factor
g
=
0.75 for an A atom in a site with
multiplicity 4 means that 75% of the site is occupied and that there are 3
A atoms in the unit cell. Obviously, the fractional population factor
cannot be greater than unity or less than zero. When the refined value is
out of the range
0
5
g
I
1,
this usually points to the incorrect
assignments of atom types or incorrectly located atom(s).
The majority of experimental powder diffraction patterns in the following
five examples were collected using a step scan data collection method on a
Scintag XDS2000 system equipped with a Ge(Li) solid-state detector cooled
with liquid nitrogen.
7.7
Completion of the model and Rietveld refinement of
NiMn02(OH)'
This example shows how to deal with complex preferred orientation, and
how to distinguish chemical elements with similar scattering factors (Ni, 28
Phase scales are treated differently in different realizations of the Rietveld algorithm, e.g.
in GSAS and in LHPM-Rietica. In the latter, the calculated absolute intensity is scaled
(normalized) to match the observed relative intensity, i.e. the scale factor is applied exactly
as shown in Eqs. 7.3 and 7.4. In the former, the observed relative intensity is scaled to
match the calculated absolute scattered intensity. In other words, the scale factors in GSAS
and LHPM-Rietica are related to one another as
KGSAS
=
lIKRietica.
R. Chen, P.Y. Zavalij, M.S. Whittingham,
J.E.
Greedan, N.P. Raju and M. Bieringer, The
hydrothermal synthesis of the new manganese and vanadium oxides, NiMn03H, MAV307
and MA0,75V4010.0.67H20 (MA
=
CH3NH3),
J.
Mater. Chem.
9,
93
(1999).

644 Chapter
7
electrons and Mn,
25
electrons) using x-ray powder data. Furthermore, in
this section we will also see how easy the latter can be done when neutron
diffraction data are available. The availability of the latter also illustrates
how to locate the hydrogen atom(s) in the unit cell. Finally, some important
geometrical aspects of the interpretation of the structural data will also be
considered.
The experimental x-ray pattern was collected in the range from
5
to
100'
28
with a
0.02O
step and a counting time of
60
seclstep. The initial model of
the crystal structure is listed in
Table
6.37.
Both independent metal atoms
are treated as manganese (Mnl and Mn2) and the hydrogen atom is missing
in this model. Thus, our goal is to distinguish between Mn and Ni atoms,
locate the H atom from the Fourier
map(s), and obtain accurate positional,
atomic displacement, and profile parameters.
Often, the initial stages of Rietveld refinement are both important and
difficult because the initial values of both the structural and profile
parameters may be far from the correct values. Hence, non-linear least
squares may be less stable when compared to the same at the end of the
refinement,
i.e. when nearly all parameters are close to their accurate values.
As mentioned above, variables should be refined in a proper order, usually
starting from only a few most critical parameters and then adding other
relevant variables, while continuously monitoring how previously refined
parameters continue to change. Those that correlate or begin to diverge
should be excluded from the refinement and, perhaps, constrained.
The following initial parameters were employed to begin this refinement:
-
Default profile parameters from the instrumental parameter file
Scintagprm
as described above in section
7.6.
-
Sample shift, S,
=
5.64
to represent sample displacements
=
-0.123
mm,
which was obtained together with the unit cell dimensions during the
lattice parameter refinement.
-
Space group
Cmc2,
and unit cell dimensions
a
=
2.8609
A,
b
=
14.6482
A,
c
=
5.2703
A, as determined earlier.
-
Structure model from
Table
6.37
with overall isotropic displacement
parameter
&,=
0.015
A2.
The starting model of the crystal structure with all of the necessary
parameters is found on the
CD
in files
Ch7ExOSa.exp1
and
Ch7E~OSa.cif;~
the experimental pattern is located in the file
Ch7ExOS-C~Ka.raw.~
'
This is the main GSAS data file, which contains all structural, instrumental and other
parameters needed for refinement.
This is a Crystallographic Information File, which records all information in a standard
format acceptable by the majority of crystallographic programs, and which is required by
the majority of technical journals for publication of the structure determination results.
'
This is the profile (histogram) file, which contains experimental powder pattern in a
standard GSAS format. Note that this format is also suitable for LHPM-Rietica.
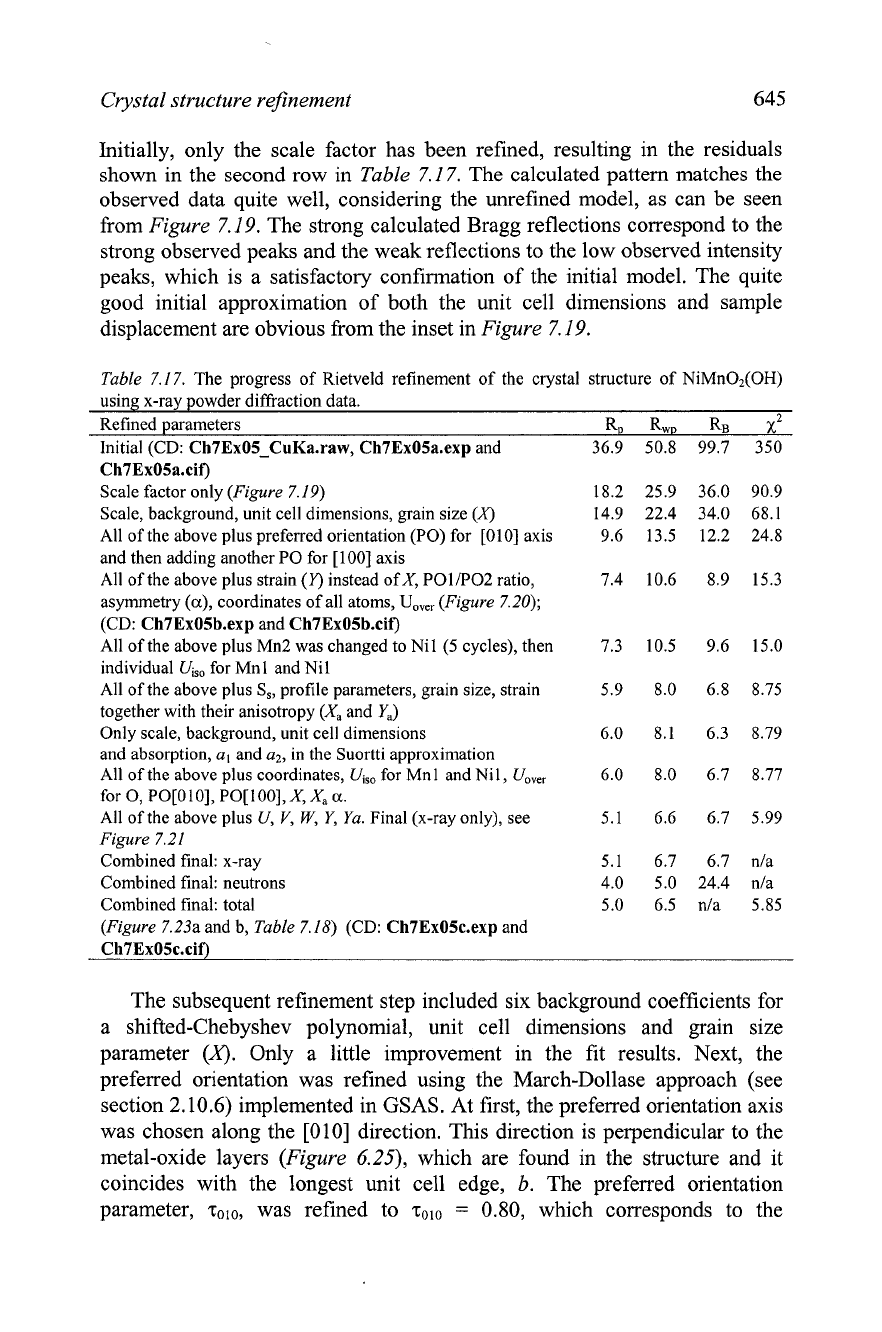
Crystal structure refinement
645
Initially, only the scale factor has been refined, resulting in the residuals
shown in the second row in
Table
7.1
7.
The calculated pattern matches the
observed data quite well, considering the unrefined model, as can be seen
from
Figure
7.19.
The strong calculated Bragg reflections correspond to the
strong observed peaks and the weak reflections to the low observed intensity
peaks, which is a satisfactory confirmation of the initial model. The quite
good initial approximation of both the unit cell dimensions and sample
displacement are obvious from the inset in
Figure
7.19.
Table 7.17. The progress of Rietveld refinement of the crystal structure of NiMnO,(OH)
using x-ray powder diffraction data.
Refined parameters
R,
RwD RB
x2
Initial (CD:
Ch7ExOS-CuKa.raw, Ch7ExOSa.exp
and
Ch7ExOSa.cif)
Scale factor only (Figure 7.19)
Scale, background, unit cell dimensions, grain size
(X)
All of the above plus preferred orientation (PO) for [010] axis
and then adding another PO for [loo] axis
All of the above plus strain
(Y)
instead ofX, P01lP02 ratio,
asymmetry (a), coordinates of all atoms,
U,,,,.
(Figure 7.20);
(CD:
Ch7ExOSb.exp
and
Ch7ExOSb.cif)
All of the above plus Mn2 was changed to Nil (5 cycles), then
individual
Ui,,
for Mnl and Nil
All of the above plus S,, profile parameters, grain size, strain
together with their anisotropy
(X,
and
Ya)
Only scale, background, unit cell dimensions
and absorption, a, and
a2,
in the Suortti approximation
All of the above plus coordinates,
Ui,,
for Mnl and Nil,
U,,,
for 0, PO[O lo], P0[100],
X,
Xa
a.
All of the above plus
U,
V,
W,
Y,
Ya. Final (x-ray only), see
Figure 7.21
Combined final: x-ray
Combined final: neutrons
Combined final: total
(Figure
7.23a and b, Table 7.18) (CD:
Ch7ExOSc.exp
and
Ch7Ex05c.cif)
The subsequent refinement step included six background coefficients for
a shifted-Chebyshev polynomial, unit cell dimensions and grain size
parameter
(3.
Only a little improvement in the fit results. Next, the
preferred orientation was refined using the March-Dollase approach (see
section 2.10.6) implemented in GSAS. At first, the preferred orientation axis
was chosen along the
[010] direction. This direction is perpendicular to the
metal-oxide layers
(Figure
6.25),
which are found in the structure and it
coincides with the longest unit cell edge,
b.
The preferred orientation
parameter, ZO~O, was refined to
z~~~
=
0.80, which corresponds to the
646
Chapter
7
preferred orientation magnitude of
2.73.'
A
second preferred orientation
axis, [loo], was added after the first, and this choice was based on the
presence of metal oxide chains along the shortest unit cell edge, a. Both
preferred orientation parameters were refined together with other variables
previously included in the least squares, resulting in a substantial lowering of
all residuals (row
4
in Table
7.1
7).
The preferred orientation axes could be
easily predicted in this structure by comparing the unit cell dimensions and
simple geometrical analysis of the model.
In
many instances, the longest unit
cell dimension (in this material the b-axis), which is perpendicular to the
layers formed in the crystal structure, is also parallel to the shortest
dimension of the plate-like crystallites.
I
R,
=
18.16%
NiMnO,(OH),
Cu
Ka
R,,
=
25.91
%
rl
10 20 30 40 50 60 70 80 90 100 110
Bragg angle,
28
(deg.)
Figure
7.19.
The observed and calculated powder diffraction patterns of NiMn02(OH) after
the initial Rietveld refinement with only the scale factor determined. The inset clarifies the
range between
70
and
90"
28.
The difference
(KObs
-
KCalc)
is shown using the same scale as
both the observed and calculated data but the plot is truncated to fit within the range
[-1500,
15001
for clarity.
'
The magnitude of the preferred orientation is the ratio between the maximum and the
minimum correction factors, which in this case, is the ratio between the correction factors
for reflections whose reciprocal lattice vectors are parallel to
d*olo
and those, which are
perpendicular to
d"olo,
i.e.
TII/Tl.
Crystal structure refinement
647
Similarly, the shortest unit cell dimension is usually parallel to the chain-
like formations in the structure (if any) and, simultaneously, to the longest
dimension of the needle-like crystallites.
In
NiMn02(OH), the a-axis is much
shorter than the two others: the needle-like crystallites are elongated along
the
[
1001
direction, with the additional preferred orientation axis
[O 101
perpendicular to the flat sides of the needles (see the inset in
Figure
6.23).
When the two preferred orientation axes are assumed, the ratio between
them should be refined as well. This was done subsequently, together with
the refinement of the coordinates of individual atoms, overall isotropic
displacement parameter,
U,,,,,
and peak asymmetry,
a.
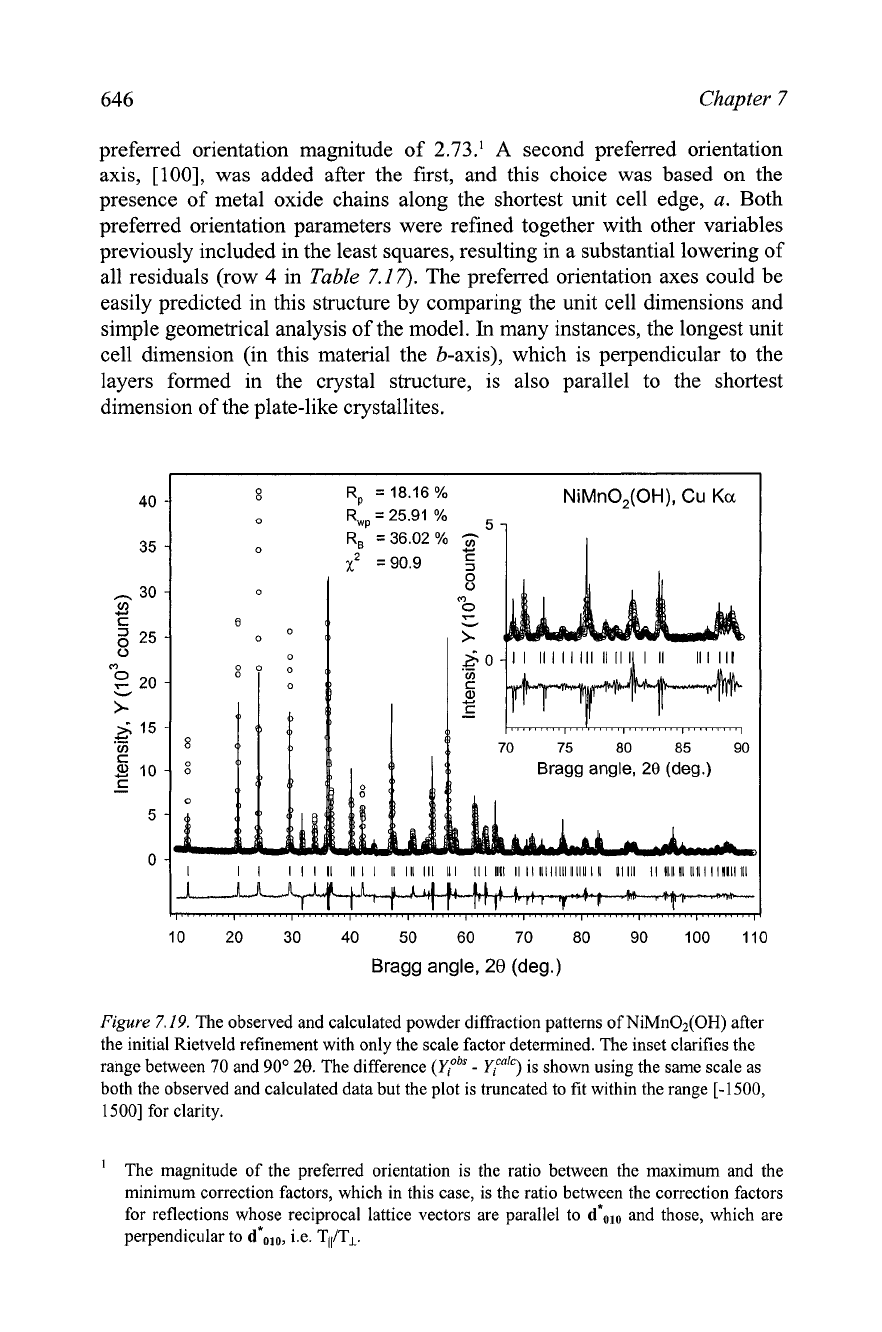
The resulting fit is
substantially improved, as shown in
Figure
7.20. It is clear, however, that
there are still some differences between the observed and calculated
intensities, as well as in the peak shapes
(e.g. see the inset in
Figure
7.20,
where some calculated peaks appear too narrow when compared with the
observed peak shapes).
NiMnO,(OH),
Cu
KCL
Bragg
angle,
20 (deg.)
10 20 30 40
50
60 70 80 90 100 110
Bragg angle,
28
(deg.)
Figure
7.20.
The observed and calculated powder diffraction patterns of NiMn02(OH) after
preferred orientation, coordinates of all atoms and the overall displacement parameter were
refined in addition to the scale factor, unit cell dimensions, background, grain size and strain
effects, and peak asymmetry. The insert clarifies the range between
70
and
90'
20.

648
Chapter
7
The next structure determination step is to distinguish the Mn and Ni
atoms in two positions, presently treated as manganese. There are several
possible ways of accomplishing this task:
-
Refining individual isotropic atomic displacement parameters with a
larger value pointing to the lighter atom and a smaller or even negative
displacement parameter indicating the heavier atom. This method works
well in refinements using single crystal diffraction data or very precise
powder data, and usually for chemical elements with substantial
differences in the scattering ability (number of electrons),
e.g. see section
7.3.3.
-
Refining site population factors: this is similar to the previous approach
but is a more appropriate way of testing for the scattering power of an
atom because the multiplication of the atomic number of the element,
currently present on a certain site, by its fractional occupation factor
results in the approximate number of electrons in the element that should
occupy the given site.
-
Conducting the refinement using all possible combinations of the
elements and then selecting the best model based on the resultant figures
of merit. This approach may be time consuming if a large number of
permutations are possible. However, in the case of NiMn02(OH), only
two possibilities exist: Mnl is Mn and Mn2 is Ni, or Mnl is Ni and Mn2
is Mn.
-
Analyzing the geometry of the model. This approach is nearly always
used in molecular compounds, even when single crystal data are
available. The analysis involves prior knowledge of possible bond
distances, angles, coordination polyhedra, etc.
-
Employing information other than Cu
Ka
x-ray diffraction data, e.g.
anomalous scattering near the K-absorption edge of one of the metals,
neutron diffraction data (see below), andlor spectroscopic results.
Despite a variety of available methods, some of them may not always
work well.
In
this case, the first three approaches based on x-ray diffraction
data did not allow clear differentiation between Mn and Ni atoms because
the differences between atomic displacement parameters, site populations
and standard figures of merit were not statistically significant. However,
quite different environments of the two independent metal atoms suggest
that a geometrical analysis may be helpful.
Thus, the bond valence sum method,' which is used mainly to
differentiate between the oxidation states of chemical elements rather than
Bond valence sum is calculated using interatomic distances and empirical bond valence
parameters tabulated for each type of the bond. The analysis was conducted using VaList
software
[A.S.
Wills and
I.D.
Brown, VaList,
CEA,
France
(1999)],
available from
ftp:Nftp.ill.frlpubldif/valist/.

Crystal structure reJinement 649
the elements themselves, was employed. The calculated bond valence sum
should be as close as possible to the oxidation state for which it was
calculated.
In
our model, the bond valence sum technique resulted in 2.93 for
the first atom, treated as ~n~', and 1.96 for the second atom, assumed to be
~i~'. The next closest possibility was 2.78 for ~i~' in the first position and
2.61 for
~n~' in the second metal site (see Table
6.37).
Therefore, bond
valence sum clearly reveals that the first metal (Mnl) is actually Mn, while
the second atom (Mn2) is Ni. Furthermore, their oxidation states are 3+ and
2+ for Mn and Ni, respectively. The final chemical composition is, therefore,
N~~+M~~+o*(oH), thus confirming the presence of a hydroxyl group in the
compound.
In
NiMn03, both Mn and Ni atoms should have oxidation states
3+ to maintain charge balance. Hence, all subsequent refinement steps
included Mn and Ni in proper sites and their atomic displacement parameters
were refined independently.
Including both grain size and strain contributions to the full width at half
maximum (X and
Y)
together with their anisotropic parts (X, and Y,)
noticeably improves the fit (Table
7.1
7).
Setting the porosity and absorption
effects using the Suortti approach as free variables (the majority of other
parameters were fixed to avoid correlations) changed the corresponding
parameters from 0.40 and 0.40 to 0.32 and
0.5 1, respectively, without
improvement of the figures of merit. Finally
U,
V, and
W
parameters, which
represent the instrumental part of the FWHM, were refined until the full
convergence was achieved. The visible improvement of the profile figures of
merit points, perhaps, to the improper preset values of
U,
V, and
W.
We note
also that X and Xa were kept fixed during the last few least squares cycles
because of their strong correlation with
Y
and Ya, and it was nonessential
which pair was refined since we had no intent to analyze grain size
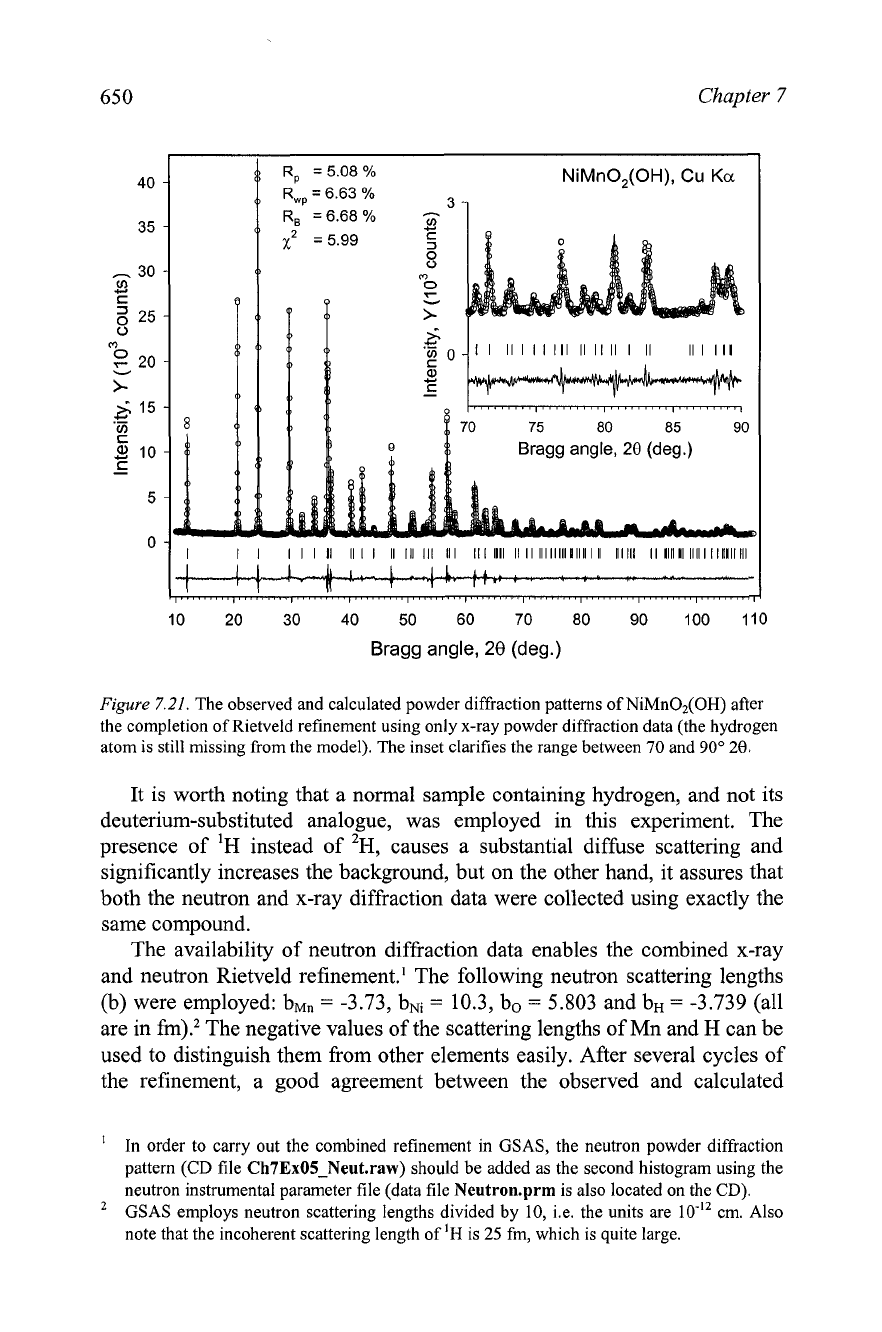
distribution and micro strain effects. The resultant observed and calculated
diffraction patterns are shown in Figure
7.21.
The results of the last refinement can be considered final if the location
of a single independent hydrogen atom in the unit is not of concern.' Not
surprisingly, it was impossible to locate hydrogen from the x-ray data
unambiguously. Therefore, we will also employ neutron powder diffraction
data collected on a powder diffractometer at the McMaster University
nuclear reactor using thermal neutrons with
h
=
1.3920
A.
'
Due to the low x-ray scattering ability of hydrogen atoms, their effect on the intensity of
powder diffraction patterns measured using conventional x-ray sources is usually
negligible, especially in the presence of relatively strongly scattering atoms, such as Mn
and Ni. Therefore, the localization of hydrogen atoms from x-ray powder diffraction data
usually presents a serious and often inexplicable problem. Hydrogen atoms positions are,
however, important in crystallography because they reveal the nature of hydrogen bonds,
which are often critical for understanding the stability of both inorganic and organic
crystals.
650
Chapter
7
R,
=
5.08
%
40
I
NiMnO,(OH),
Cu
Ka
R,,
=
6.63
%
m
.-
4-
0-1
I I1 I I1 Ill I1 llll I I1 Ill Ill
a
C
8
-
.-
V,
75
80
85
90
B
10
Bragg
angle,
28
(deg.)
n
10 20 30 40 50
60
70
80 90
100 110
Bragg angle, 28
(deg.)
Figure
7.21.
The observed and calculated powder diffraction patterns of NiMn02(OH) after
the completion of Rietveld refinement using only x-ray powder diffraction data (the hydrogen
atom is still missing from the model). The inset clarifies the range between
70
and
90"
20.
It is worth noting that a normal sample containing hydrogen, and not its
deuterium-substituted analogue, was employed in this experiment. The
presence of
'H
instead of 2~, causes a substantial diffuse scattering and
significantly increases the background, but on the other hand, it assures that
both the neutron and x-ray diffraction data were collected using exactly the
same compound.
The availability of neutron diffraction data enables the combined x-ray
and neutron Rietveld refinement.' The following neutron scattering lengths
(b) were employed: bM,
=
-3.73, bNi
=
10.3, bo
=
5.803 and bH
=
-3.739 (all
are in fm).2 The negative values of the scattering lengths of Mn and H can be
used to distinguish them from other elements easily. After several cycles of
the refinement, a good agreement between the observed and calculated
'
In order to carry out the combined refinement in GSAS, the neutron powder diffraction
pattern
(CD
file Ch7ExOS-Neut.raw) should be added as the second histogram using the
neutron instrumental parameter file (data file Neutron.prm is also located on the
CD).
GSAS employs neutron scattering lengths divided by
10,
i.e. the units are
10-12
cm. Also
note that the incoherent scattering length of
'H
is
25
fm, which is quite large.

Crystal structure refinement
65
1
patterns was achieved. This result decisively proves that the Mn and Ni
positions were recognized accurately: their scattering factors have opposite
signs and, when switched, the computed intensities will be quite different.
The difference Fourier map was calculated using the neutron data and it
is shown in
Figure
7.22a. The position of an
H
atom can be found as the
deepest minimum on the map. Yet this minimum is only slightly deeper than
other extremes, all of which could be considered as noise, which appears due
to the relatively low accuracy of the experimental data. The confirmation of
the
H
position was made by using chemical intuition considering the crystal
structure model, especially because the geometry of the hydrogen bond
01-
H.e.03
is nearly ideal. Positions of all atoms in the model have been
confirmed on the conventional Fourier map, which is shown in
Figure
7.22b.
Figure
7.22.
Distributions of the nuclear density in the unit cell of NiMn02(OH) at
x
=
0:
a) the difference Fourier map calculated using IAFI
=
IFobs-Fcalcl and phase angles calculated
without
H
atom (pmi,
=
-2.4
fm/A3, pmax
=
2.4
fm/A3, Ap
=
0.4
fm/A3, p(H)
=
-2.4
fm/A3);
b)
the conventional Fourier map computed using IFob,/ and phase angles calculated including the
H
atom (pmi,
=
-12
fm/A3, pmax
=
18
fm/A3 (high p on Ni atoms is not shown), Ap
=
2
fm/A3,
p(H)
=
-10
fm/A3, p(Mn)
=
-12
fm/A3). Solid lines show positive values, thin dotted lines
negative, and thick dotted lines indicate zero level.