Ojima I. (ed.) Fluorine in Medicinal Chemistry and Chemical Biology
Подождите немного. Документ загружается.

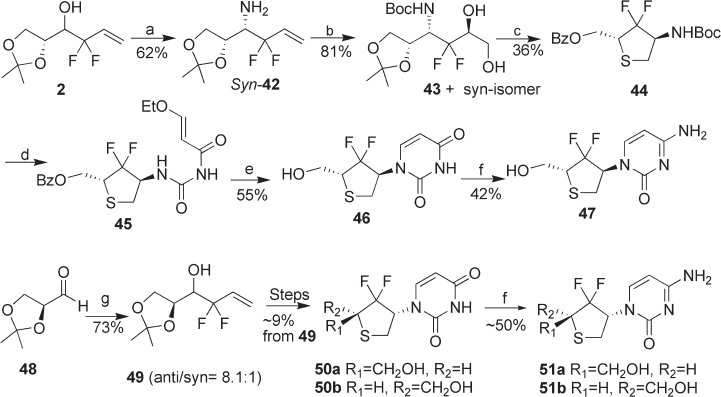
Synthesis of gem-Difl uoromethylenated Nucleosides 207
Boc group of 44 , followed by the condensation of the resultant amine with 3 - ethoxy - 2 -
propenoyl isocyanate, afforded urea 45 . The cyclization of 45 with sulfuric acid and the
subsequent removal of the benzoyl group gave thionucleoside 46 . Nucleoside 46 was
further converted to thionucleoside 47 . Meanwhile, gem - difl uorohomoallyl alcohol 49 ,
which was derived from ( S ) - glyceraldehyde acetonide 48 and 3 - bromo - 3,3 - difl uoropro-
pene, was converted to thionucleosides 50a and 50b by applying the same strategy [13] .
Thionucleosides 50a and 50b were further converted to thionucleosides 51a and 51b (see
Scheme 8.7 ).
8.3 Synthesis of gem - Difl uoromethylenated Azanucleosides via
Difl uoromethylenated l - Proline Derivatives
Azanucleosides, containing a nitrogen atom instead of the oxygen atom on the sugar ring,
possess unique biological properties as the nitrogen atom not only has the heteroatom
effect but also can bind strongly to certain DNA repair enzymes [14] . During the course
of our study on fl uorinated amino acids, a versatile procedure was developed for the
preparation of 2 ′ ,3 ′ - dideoxy - 2 ′ - difl uoromethylazanucleosides (see Scheme 8.8 ) [15] . Natu-
rally occurring trans - 4 - hydroxy - l - proline was converted to difl uoro - olefi n 52 in 30% yield
via Swern oxidation, followed by difl uoromethylenation of the resultant ketone with
CF
2
Br
2
/Zn/HMPT2. Olefi n 52 was reduced to give 53 as a 7 : 1 cis / trans mixture via
Scheme 8.7 Reagents and conditions: (a) (i) Tf
2
O, pyridine; (ii) NaN
3
, DMF; (iii) PPh
3
, THF,
(iv) H
2
O; (b) (i) Boc
2
O; (ii) OsO
4
, NMMO; (c) (i) AcOH; (ii) NaIO
4
; (iii) NaBH
4
/MeOH;
(iv) MsCl, pyridine; (v) Na
2
S, DMF; (d) (i) TFA; (ii) 3 - ethoxy - 2 - propenoyl isocyanate; (e) (i)
2 M H
2
SO
4
; (ii) sat. NH
3
/MeOH; (f) (i) Ac
2
O, DMAP; (ii) TPSCl, DMAP, Et
3
N; (iii) conc.
NH
3
· H
2
O; (g) In powder, CH
2
=
CHCF
2
Br, DMF.

208 Fluorine in Medicinal Chemistry and Chemical Biology
catalytic hydrogenation. After oxidation of the 5 - methylene group of 53 , pyrrolidone 54a
was obtained in 68% yield along with its trans - isomer 54b . After separation, 54a was
converted to 55 by reduction and silyl protection. Protection of the 1 - amino group of 55
with a benzyloxycarbonyl group gave 56 , which was converted to acetate 57 by reduction
and acetylation. Acetate 57 was then coupled with silylated uracil or thymine to furnish
azanucleosides, 58a/b or 59a/b , as a mixture of α - and β - anomers (see Scheme 8.8 ). The
absolute confi guration of compound 59b was confi rmed by x - ray crystallographic
analysis.
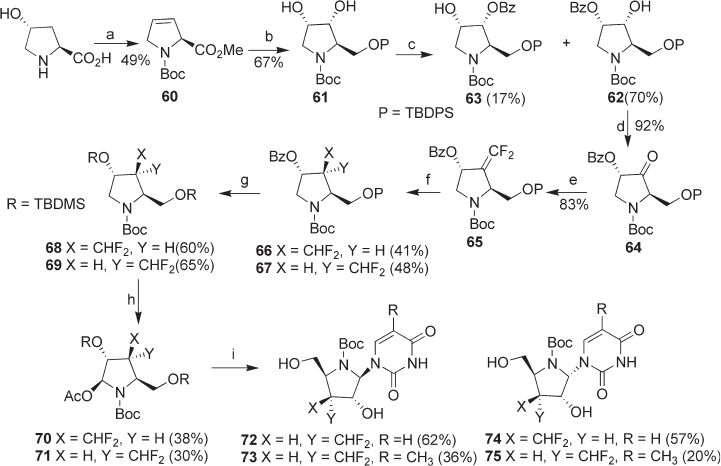
Another strategy to synthesize 3 ′ - difl uoromethylated azanucleosides 72 – 75 , starting
from trans - 4 - hydroxy - l - proline, was also developed (see Scheme 8.9 ) [16] . Diol 61 was
obtained in a straightforward manner by dihydroxylation of 60 , which was derived from
trans - 4 - hydroxy - l - proline. Mono - benzoylation of diol 61 gave 62 as the main product
(70%) along with compound 63 (17%). Compound 62 was then treated with Dess – Martin
reagent to afford ketone 64 , which was converted to difl uoromethylene - pyrrolidine 65 by
reacting with CF
2
Br
2
/Zn/HMPT. Catalytic hydrogenation of 65 gave a separable mixture
of 66 and 67 . Compound 66 was the main product when 10%Pd/C was used, whereas
compound 67 was the major product with Pd(OH)
2
/C. However, the attempted oxidation
of the methylene group of 66 or 67 using RuO
2
· x H
2
O/NaIO
4
failed to give the desired
pyrrolidinones, probably due to the existence of tert - butyldiphenylsilyl group. Thus, the
protecting groups of 66 and 67 were changed to tert - butyldimethylsilyl groups not only
for the hydroxymethyl group at C - 2, but also for the secondary hydroxyl group at C - 4 to
afford the corresponding 68 and 69 , respectively. Then, 68 and 69 were subjected to a
series of transformations similar to those described for the synthesis of 58 and 59 from
53 (see Scheme 8.8 ) to give 3 ′ - difl uoromethylated azanucleosides 72 , 73 , 74 and 75 (see
Scheme 8.9 ).
Scheme 8.8 Reagents and conditions: (a) (i) SOCl
2
, MeOH; (ii) Boc
2
O, Et
3
N, DMAP,
CH
2
Cl
2
; (iii) Swern oxidation; (iv) CF
2
Br
2
/Zn/HMPT; (b) Pd/C, H
2
, EtOH, r.t., 1 atm; (c)
RuO
2
· x H
2
O, NaIO
4
, EtOAc, H
2
O, r.t.; (d) (i) TFA, CH
2
Cl
2
; (ii) NaBH
4
, MeOH, − 78 ° C – 0 ° C;
(iii) TBDMSCl, imidazole, DMAP, CH
2
Cl
2
; (e) LHMDS, CbzCl, THF, − 78 ° C; (f) (i) LiBEt
3
H,
THF, − 78 ° C; (ii) Ac
2
O, DMAP, Py; (g) (i) silylated uracil or silylated thymine, TMSOTf,
MeCN; (ii) TBAF, THF.
Synthesis of gem-Difl uoromethylenated Nucleosides 209
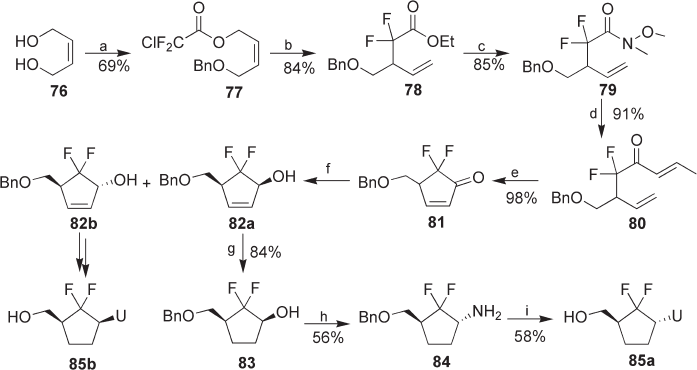
8.4 Synthesis of gem - Difl uoromethylenated Carbocyclic Nucleosides
In recent years, increasing attention has been paid to the structural modifi cations of car-
bocyclic nucleosides. Because of the absence of glycosidic linkage, carbocyclic nucleo-
sides are chemically more stable and not vulnerable to phosphorylases that cleave the
N - glycoside linkage in usual nucleosides [17] . The fi rst synthesis of gem - difl uorometh-
ylenated carbocyclic nucleosides was reported by Borthwick et al. in 1990 via direct
difl uorination of a ketone moiety on a carbocyclic ring with DAST [18] . Recently, 2 ′ ,3 ′ -
dideoxy - 6 ′ ,6 ′ - difl uorouracils, 85a and 85b , were synthesized using a new strategy in 14
steps starting from ( Z ) - 2 - butene - 1,4 - diol ( 76 ) (see Scheme 8.10 ) [19] . The new strategy
included the construction of a carbocyclic ring via Reformatskii – Claisen rearrangement
and ring - closing metathesis. ( Z ) - 2 - Butene - 1,4 - diol ( 76 ) was converted to chlorodifl uoro-
acetate 77 in 69% yield, which was subjected to a silicon - induced Reformatskii – Claisen
rearrangement to give difl uoro ester 78 . Ester 78 was then transformed to Weinreb amide
Scheme 8.9 Reagents and conditions: (a) (i) SOCl
2
, MeOH; (ii) Boc
2
O, Et
3
N, DMAP,
CH
2
Cl
2
; (iii) MsCl, Et
3
N, DMAP, CH
2
Cl
2
, r.t.; (iv) PhSeSePh, MeOH, refl ux; v. H
2
O
2
, Py, r.t.;
(b) (i) LiAlH
4
, Et
2
O, r.t.; (ii) TBDPSCl, imidazole, DMAP, CH
2
Cl
2
, r.t.; (iii) OsO
4
, NMNO,
acetone/H
2
O, r.t.; (c) BzCl, Py, CH
2
Cl
2
, − 10 ° C, 24 h; (d) (i) BzCl, Py, CH
2
Cl
2
, − 10 ° C, 24 h;
(ii) Dess – Martin oxidant, CH
2
Cl
2
, r.t.; (e) CF
2
Br
2
, HMPT, Zn, THF, refl ux; (f) 10% Pd/C,
70 atm, or Pd(OH)
2
/C, H
2
, 80 atm; (g) (i) TBAF, THF, r.t.; (ii) TBDMSCl, imidazole, DMAP,
CH
2
Cl
2
, r.t.; (iii) sat. NH
3
/MeOH, r.t.; (iv) TBDMSCl, imidazole, DMAP, DMF; (h) (i)
RuO
2
· x H
2
O, NaIO
4
, EtOAc, H
2
O, r.t.; (ii) LiBEt
3
H, THF, − 78 ° C; (iii) Ac
2
O, CH
2
Cl
2
, Et
3
N,
DMAP, r.t.; (i) (i) silylated uracil or thymine, N,O - bis(trimethylsilyl)acetamide, TMSOTf, 0 ° C
to r.t.; (ii) TBAF, THF, r.t.
210 Fluorine in Medicinal Chemistry and Chemical Biology
79 and treated with allylmagnesium bromide to afford diene 80 via a double - bond isom-
erization. Diene 80 was subjected to ring - closing metathesis. However, when 80 was
treated with the fi rst - generation Grubbs catalyst, the reaction did not occur. The second -
generation Grubbs catalyst was therefore employed for the reaction and the reaction pro-
ceeded smoothly to give cyclopentenone 81 in nearly quantitative yield. Luche reduction
of 81 afforded a 2.9 : 1 separable mixture of unsaturated alcohols, 82a and 82b . Catalytic
hydrogenation of 82a gave alcohol 83 , which was transformed to α - 2 ′ ,3 ′ - dideoxy - 6 ′ ,6 ′ -
difl uorocarbocyclic uridine 85a . The relative stereochemistry of 85a was determined by
x - ray crystallographic analysis. In the same manner, unsaturated alcohol 82b was con-
verted to β - anomer 85b . Thus, racemic 2 ′ ,3 ′ - dideoxy - 6 ′ ,6 ′ - difl uorocarbocyclic uracils, 85a
and 85b , were obtained in 14 steps from 76 in 7.6% and 1.5% overall yields, respectively
(see Scheme 8.10 ).
8.5 Conclusion
gem - Difl uoromethylenated nucleosides constitute important members of the extensively
studied nucleoside analogues, which are an important class of candidates for new antiviral
and antitumor agents. We have described here recent development in the syntheses of
difl uoromethylenated nucleosides in our laboratory. With the development of more practi-
Scheme 8.10 Reagents and conditions: (a) (i) NaH, DMF, BnBr, 0 ° C; (ii) ClCF
2
CO
2
H, cat.
H
2
SO
4
, toluene, 140 ° C; (b) (i) zinc dust, TMSCl, CH
3
CN, 100 ° C; (ii) ClCF
2
CO
2
H, cat. H
2
SO
4
,
toluene, 140 ° C; (b) (i) zinc dust, TMSCl, CH
3
CN, 100 ° C; (ii) cat. H
2
SO
4
, EtOH, 40 ° C; (c)
N , O - dimethylhydroxylamine, AlMe
3
, toluene; (d) (i) CH
2
=
CHCH
2
MgBr, THF; (ii) Et
3
N, THF;
(e) Grubbs ’ II catalyst, toluene, 80 ° C; (f) CeCl
3
· 7H
2
O, NaBH
4
, 0 ° C; (g) H
2
, Pd black,
benzene; (h) (i) Tf
2
O, pyridine, CH
2
Cl
2
, − 40 ° C; (ii) NaN
3
, DMF; (iii) H
2
, cat. Pd black,
benzene; (i) (i) ( E ) - EtOCH
=
CHCONCO, DMF, − 25 ° C; (ii) 2 N H
2
SO
4
, refl ux; (iii) H
2
, cat.
Pd/C.
Synthesis of gem-Difl uoromethylenated Nucleosides 211
cal difl uoromethylenation methodologies and more knowledge of structure – activity rela-
tionships, more effi cacious gem - difl uoromethylenated nucleosides will be developed as
novel antiviral and antitumor agents.
Acknowledgment
We are grateful to the National Natural Science Foundation of China, the Ministry of
Education of China and Shanghai Municipal Scientifi c Committee for the fi nancial
support.
References
For recent fl uorinated nucleosides reviews, see (a) Meng , W - D. and Qing F - L. ( 2006 ) Fluori-
nated nucleosides as antiviral and antitumor agents . Current Topics in Medicinal Chemistry , 6 ,
1499 – 1528 ; (b) Qing , F - L. and Qiu , X - L. ( 2007 ) Synthesis of gem - difl uoromethylated sugar
nucleosides . In: Current Fluoroorganic Chemistry , ACS Symposium Series 949 , pp. 305 – 322
and references cited therein.
Blackburn , G. W. , England , D. E. and Kolmann , F. ( 1981 ) Monofl uoro - and difl uoro -
methylenebisphosphonic acids: isopolar analogues of pyrophosphoric acid . J. Chem. Soc.,
Chem. Commun. , 930 – 932 .
(a) Lee , D. ( 2003 ) Gemcitabine - based therapy in relapsed non - Hodgkin ’ s lymphoma and cutane-
ous T - cell lymphoma . Clin Lymphoma Myeloma , 4 , 152 – 153 ; (b) Nobel , S. and Goa , K. L.
( 1997 ) Gemcitabine. A review of its pharmacology and clinical potential in non - small cell lung
cancer and pancreatic cancer . Drugs , 54 , 447 – 472 .
Zhang , X. G. , Xia , H. R. , Dong , X. C. , et al. ( 2003 ) 3 - Deoxy - 3,3 - difl uoro - d - arabinofuranose:
First stereoselective synthesis and application of gem - difl uorinated sugar nucleosides . J. Org.
Chem. , 68 , 9026 – 9033 .
Xu , X. H. , Qiu , X. L. , Zhang , X. G. and Qing , F. L. ( 2006 ) Synthesis of l - and d - β - 3 ′ - deoxy -
3 ′ ,3 ′ - difl uoronucleosides . J. Org. Chem. , 71 , 2820 – 2824 .
Vorbr ü ggen , H. and Hofl e , G. ( 1981 ) Nucleoside syntheses XXIII: On the mechanism of nucleo-
side synthesis . Chem. Ber. , 114 , 1256 – 1268 .
Vorbr ü ggen , H , Krolikiewicz , K. and Bennua. B. ( 1981 ) Nucleoside syntheses XXII: Nucleoside
synthesis with trimethylsilyl trifl ate and perchlorate as catalysts . Chem. Ber. 114 , 1234 – 1256 .
(a) Barrena , M. I. , Matheu , M. I. and Castillon , S. ( 1998 ) An improved synthesis of 4 - O - benzoyl -
2,2 - difl uorooleandrose from l - rhamnose. Factors determining the synthesis of 2,2 - difl uorocar-
bohydrates from 2 - uloses . J. Org. Chem. , 63 , 2184 – 2188 . (b) Aghmiz , M. L. , Diaz , Y. , Jana ,
G. H. , et al. ( 2001 ) The reaction of pyranoside 2 - uloses with DAST revised. Synthesis of 1 -
fl uoro - ketofuranosyl fl uorides and their reactivity with alcohols . Tetrahedron , 57 , 6733 – 6743 .
Xu , X. H. , You , Z. W. , Zhang , X. G. and Qing , F. L. ( 2007 ) Synthesis of 3 - deoxy - 3,3 - difl uoro -
d - ribohexose from gem - difl uorohomoallyl alcohol . J. Fluorine Chem. , 128 , 535 – 539 .
For thionucleosides reviews, see: Yokoyama , M. ( 2000 ) Synthesis and biological activity of
thionucleosides . Synthesis , 1637 – 1655 .
Zheng , F. , Zhang , X. H. , Qiu , X. L. , et al. ( 2006 ) Synthesis of l - β - 3 ′ - deoxy - 3 ′ ,3 ′ - difl uoro - 4 ′ -
thionucleosides . Org. Lett. , 8 , 6083 – 6086 .
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
212 Fluorine in Medicinal Chemistry and Chemical Biology
Wu , Y. Y. , Zhang , X. , Meng , W. - D. and Qing , F. - L. ( 2004 ) Synthesis of new 2 ′ ,3 ′ - dideoxy -
6 ′ ,6 ′ - difl uoro - 3 ′ - thionucleoside from gem - difl uorohomoallyl alcohol . Org. Lett. , 6 ,
3941 – 3944 .
Yue , X. , Wu , Y. Y. and Qing , F. - L. ( 2007 ) Synthesis of a series of novel 2 ′ ,3 ′ - dideoxy - 6 ′ ,6 ′ -
difl uoro - 3 ′ - thionucleosides . Tetrahedron , 63 , 1560 – 1567 .
For azanucleosides reviews, see: Yokoyama , M. and Momotake , A. ( 1999 ) Synthesis and bio-
logical activity of azanucleosides . Synthesis , 1541 – 1554 .
Qiu , X. L. and Qing , F. - L. ( 2004 ) Synthesis of 2 ′ ,3 ′ - dideoxy - 2 ′ - difl uoromethyl azanucleosides .
Synthesis , 334 – 340 .
Qiu , X. L. and Qing , F. - L. ( 2005 ) Synthesis of 3 ′ - dideoxy - 3 ′ - difl uoromethyl azanucleosides
from trans - 4 - hydroxy - l - proline . J. Org. Chem. , 70 , 3826 – 3837 .
For carbocyclic nucleosides reviews, see: (a) Crimmins , M. T. ( 1998 ) New developments in the
enantioselective synthesis of cyclopentyl carbocyclic nucleosides . Tetrahedron , 54 , 9229 – 9272 .
(b) Borthwick , A. D. and Biggadike , K. ( 1992 ) Synthesis of chiral carbocyclic nucleosides .
Tetrahedron , 48 , 571 – 623 . (c) Agrofoglio , L. , Suhas , E. , Farese , A. , et al. ( 1994 ) Synthesis of
carbocyclic nucleosides . Tetrahedron , 50 , 10611 – 10670 . (d) Marquez , V. E. and Lim , M. - U.
( 1986 ) Med. Res. Rev. , Carbocyclic nucleosides 6 , 1 – 40 . (e) Wu , Q. and Simons , C. ( 2004 )
Synthetic methodologies for C - nucleosides . Synthesis , 1533 – 1553 . (f) Wang , J. , Froeyen , M.
and Herdewijn , P. ( 2004 ) Six - membered carbocyclic nucleosides . Adv. Antivir. Drug Des. , 4 .
119 – 145 . (g) Rodriguez , J. B. and Comin , M. J. ( 2003 ) New progresses in the enantioselective
synthesis and biological properties of carbocyclic nucleosides Mini Rev . Med. Chem. , 3 ,
95 – 114 .
Brothwick , A. D. , Evans , D. N. , Kirk , B. E. , et al. ( 1990 ) Fluoro carbocyclic nucleosides: syn-
thesis and antiviral activity of 2 ′ - and 6 ′ - fl uoro carbocyclic pyrimidine nucleosides including
carbocyclic 1 - (2 - deoxy - 2 - fl uoro - β - d - arabinofuranosyl) - 5 - methyluracil and carbocyclic 1 - (2 ′ -
deoxy - 2 - fl uoro - β - d - arabinofuranosyl)5 - iodouracil . J. Med. Chem. , 33 , 179 – 186 .
Yang , Y. Y. , Meng W. - D. and Qing , F. - L. ( 2004 ) Synthesis of 2 ′ ,3 ′ - dideoxy - 6 ′ ,6 ′ - difl uorocar-
bocyclic nucleosides . Org. Lett. , 6 , 4257 – 4259 .
12.
13.
14.
15.
16.
17.
18.
19.

9
Recent Advances in the Syntheses of
Fluorinated Amino Acids
Kenji Uneyama
9.1 Introduction
Fluorinated amino acids have been used as components of modifi ed peptides and proteins
in protein engineering [1] and have also found applications as potential enzyme inhibi-
tors and antitumor and antibacterial agents [2 – 4] . To date, due to the potent biological
activities of fl uorinated α - amino acids, several synthetic strategies and methods have
been the subjects of reviews and books [5, 6] . In this chapter synthetic strategies for
fl uorinated amino acids recently developed in the 2000s are summarized. Syntheses of
fl uorinated amino acids are classifi ed into three groups on the basis of the strategies for
creation of the chiral center: (1) enantioselective, (2) diastereoselective, and (3) racemic
syntheses.
9.2 Enantioselective Synthesis
Enantioselective synthesis involves chemical modifi cation via (section 9.2.1 ) introduction of
a chiral center into achiral fl uorinated building blocks, (9.2.2) chiral transposition, (9.2.3)
introduction of fl uorine functionality into nonfl uorinated chiral building blocks, (9.2.4) mod-
ifi cation of chiral fl uorinated building blocks, and (9.2.5) enzymatic resolution of racemic
fl uorinated building blocks. The following sections summarize these fi ve categories.
Fluorine in Medicinal Chemistry and Chemical Biology Edited by Iwao Ojima
© 2009 Blackwell Publishing, Ltd. ISBN: 978-1-405-16720-8
214 Fluorine in Medicinal Chemistry and Chemical Biology
9.2.1 Introduction of a Chiral Center into Achiral Fluorinated Building Blocks
9.2.1.1 Introduction of Chirality by Hydrogenation
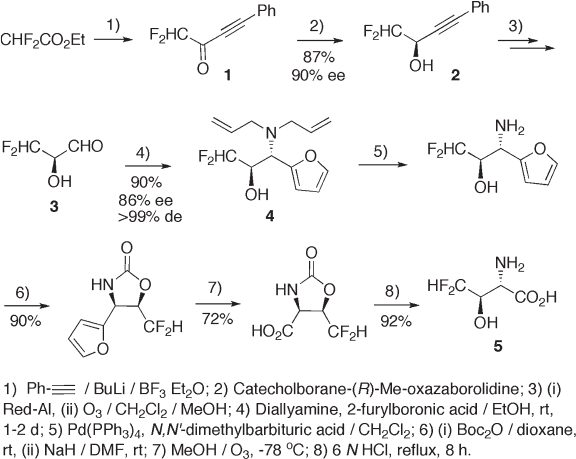
Asymmetric hydrogenation of either a carbonyl or an imino group to a hydroxyl group or
an amino group has frequently been employed for the introduction of chirality in amino
acid syntheses. Corey ’ s catecolborane – oxazaborolidine protocol enables transformation of
difl uoromethyl ketone 1 into alcohol 2 with excellent enantioselectivity. The reaction of
diastereoselective amination of α - hydroxyaldehyde 3 with N , N - diallylamine and 2 - furyl-
boronic acid provides furyl amino alcohol 4 in good chemical yield along with excellent
diastereoselectivity. This protocol is applicable for the preparation of amino acids and
amino alcohols with a trifl uoromethyl group by the combination of N , N - diallyl or N , N -
dibenzyl amine and aromatic, heteroaromatic and alkenyl boronic acids [7] . The usual
chemical transformations as shown in steps 5 to 8 in Scheme 9.1 lead to (2 S ,3 R ) -
difl uorothreonine 5 [8] .
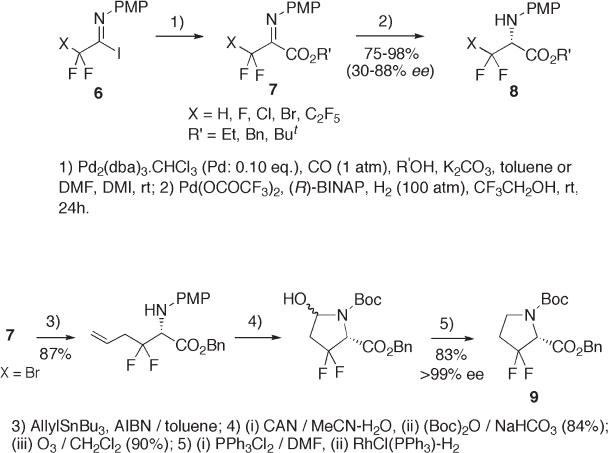
Palladium - catalyzed carboalkoxylation of imidoyl iodides 6 provides benzyl [9] and
even tert - butyl [10] esters 7 . Asymmetric hydrogenation of the imino moiety of imono
esters 7 in a Pd(OCOCF
3
)
2
/ ( R ) - BINAP / CF
3
CH
2
OH system gives enantio - enriched amino
esters 8 in 85 – 91% ee (see Scheme 9.2 ) [11] .The enantioselectivity achieved by the hydro-
genation was much better than that by Corey ’ s hydride reduction [12] and was employed
for the syntheses of enantiomerically pure N - Boc - β , β - difl uoroproline benzyl ester 9 (see
Scheme 9.3 ) [13] and enantiomerically enriched N - Boc - β , β - difl uoroglutamic acid benzyl
ester [13] .
Scheme 9.1
Recent Advances in the Syntheses of Fluorinated Amino Acids 215
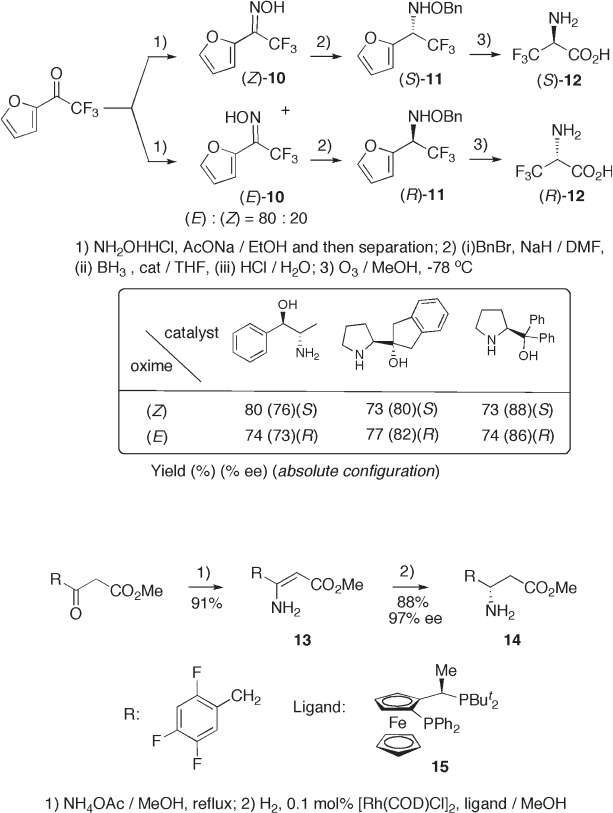
Interestingly, the stereochemistry of the hydroxyl group on the imino nitrogen in
oxime 10 affects the stereochemistry of hydroxylamine 11 (see Scheme 9.4 ). Thus, hydride
reduction of either ( Z ) - or ( E ) - oximes 10 with hydroborane in the presence of chiral amino
alcohols produces ( S ) - and ( R ) - N - benzyl oximes 11 , respectively, as shown in the table in
Scheme 9.4 , which were subsequently transformed to ( R ) - and ( S ) - trifl uoroalanine 12
[14] .
The carbon – carbon double bond of an enamine is also applicable for asymmetric
hydrogenation leading to chiral amino acids. For example, hydrogenation of 13 by rhodium
catalyst with ferrocenyl diphosphine 15 as a ligand was successful for the synthesis of
methyl 3 - amino - 4 - polyfl uorophenylbutanoate 14 with excellent stereoselectivity (see
Scheme 9.5 ) [15] .
9.2.1.2 Asymmetric Dihydroxylation
Asymmetric dihydroxylation of trifl uoromethylalkenes is also useful for construction of
enantio - enriched trifl uoromethylated diols usable for trifl uoromethylated amino acids with
chiral hydroxyl group. Thus, Sharpless AD reaction of 16 provides diol 17 with excellent
enantioselectivity. Regioselective and stereospecifi c replacement of the sulfonate moiety
in 18 with azide ion enables the introduction of nitrogen functionality. A series of well -
known chemical transformation of 19 leads to 4,4,4 - trifl uorothreonine 20 (see Scheme 9.6 )
[16] . Dehydroxylative - hydrogenation of 21 by radical reaction via thiocarbonate and sub-
sequent chemical transformation synthesize enantio - enriched ( S ) - 2 - amino - 4,4,4 - trifl uoro-
butanoic acid 22 [16] . Both enantiomers of 20 and 22 were prepared in a similar manner
from (2 R ,3 S ) - diol of 17 .
Scheme 9.2
Scheme 9.3
216 Fluorine in Medicinal Chemistry and Chemical Biology
Scheme 9.4
Scheme 9.5
9.2.1.3 Asymmetric Alkylation
Asymmetric alkylation of imines has been employed most frequently for the construction
of chiral amino moieties involved in the syntheses of nitrogen heterocycles and amino
acids [17] . This approach is also useful for fl uorinated amino acid synthesis as shown in
Scheme 9.7 . Mannich reaction of enolate with imino ester 23 in the presence of L - proline
gives α - amino esters 24 and 25 enantio - and diastereoselectively [18] .
Asymmetric alkylation of N - protected glycine ester 26 under phase - transfer catalysis
conditions is the well - known method for the syntheses of α - amino acids [19] . Scheme