Jackson S.D., Hargreaves J.S.J. Metal Oxide Catalysis
Подождите немного. Документ загружается.


8.4 Calculation of Surface Structure 375
Figure 8.21 c and d shows the density of states decomposed by layer for the fi rst
three surface planes. At the surface the octahedral environment of the bulk Ni
2+
ions is lost and so the e
g
orbitals are no longer degenerate. This is seen as a split-
ting of the minority spin states at the bottom of the conduction band in the PW91
calculation with the d
z
2
orbital ( z perpendicular to the surface) moving to lower
energies, giving a further reduction in the estimated e
g
to just 0.1 eV. A similar
effect is seen for the PW91 + U model, which gives a surface layer band gap of
2.9 eV. By the second sub - surface layer the symmetry of the cation sites appears
to be O
h
as the splitting is removed.
The adsorption of both CO and NO on the NiO(100) surface has been investi-
gated using photoelectron diffraction [115] giving a tilted geometry with Ni
–
C and
Ni
–
N distances signifi cantly longer than predicted using gradient - corrected DFT.
Rohrbach and coworkers showed that the PW91 + U approach is able to give much
better agreement with the experimental data for CO and NO. [116] . The observed
tilted geometries are unexpected from comparable metal complex chemistry, for
which π back - donation from metal to ligand would favor a linear geometry.
However, in the metal oxide surface we have seen how exchange splitting shifts
the occupied metal d orbital bands to lower energies and so their mixing with
adsorbate orbitals becomes less effi cient in a PW91 + U model. Molecular tilting
allows σ donation of charge from the diatomic to the surface d
z
2
from both the
5 σ and occupied π orbitals, which would be symmetry forbidden in an upright
adsorption mode. For the NO case, the partially occupied 2 π * orbital is hybridized
with the d
z
2
and the 5 σ contribution is almost negligible. Similar geometries for
NO adsorption on MgO, where there is no option for back - donation, have been
found in the PW91 calculations of Schneider and coworkers [117] .
The use of HF - like terms in Equation 8.40 suggests that just using HF itself for
these types of material may be preferable. Indeed Towler and coworkers have
shown that UHF correctly predicts the anti - ferromagnetic state to be lower in
energy than either the ferromagnetic state or a solution with spins paired on a
site - by - site basis. However, their estimated band gap of around 13.6 eV is a signifi -
cant overestimate of the experimental value of 3.8 eV [58] . A much better agree-
ment between calculated and experimental band gaps can be obtained with the
hybrid B3LYP function, which gives 3.9 eV in this case [57] .
Indeed, in these problems involving electron localization, hybrid functionals
provide an alternative approach that has the advantage that exchange is applied
uniformly to the electron density and not just to the transition metal centers. In
the next section we look at examples using either DFT + U or hybrid functionals
to examine defect states on oxide surfaces.
8.4.5
Defects on the Surfaces of Transition Metal Oxides
Anion defects on oxide surfaces are important centers for adsorption and activa-
tion of molecules in catalysis. For example, oxidation reactions following a
Mars – van Krevelen mechanism require oxygen transfer from a surface to an

376 8 Theory: Periodic Electronic Structure Calculations
adsorbate, a process that creates an anion defect. Reduction of a metal oxide in its
simplest form can be thought of as the liberation of oxygen:
MO MO e O g
mn mn
=++
()
−12
2
1
2
(8.42)
In a calculation, m and n defi ne the stoichiometry of the simulation cell, and
the two electrons that remain after a surface O
2 −
ion has formed part of the neutral
O
2
(g) molecule have been written explicitly. Using a periodic approach it should
be remembered that the defect will be repeated in neighboring cells. To obtain
results relevant to isolated defect sites requires cell sizes large enough to allow
interactions between the defect and its own images to be neglected.
In alkaline earth oxides such as MgO, the two electrons of Equation 8.42 are
known to be localized at the defect site by the Madelung potential of the solid [55] .
However, in reducible oxides such as TiO
2
there is the alternative of localizing the
electrons at metal sites to give two Ti
3+
centers as shown schematically in Figure
8.22 . Reduction of a stoichiometric oxide in this way is a local event giving an
electronic structure in which the reduced metal centers are mixed in with fully
oxidized ions. As we have seen, the correct modeling of charge and spin states in
such a system requires a balanced treatment of exchange and correlation energies
and is currently best achieved using either hybrid functionals or a DFT + U
scheme.
Figure 8.22 also shows how heterolytic cleavage of water at such a defect site
appears to repair the structure of the surface but leaves the two metal sites reduced.
For the resulting hydroxylated surface it is possible to push electrons around to re -
oxidize one center, creating an alternative description of the surface with a hydroxy
radical. EELS experiments show little difference between the defective and hydroxyl-
ated surfaces [118] suggesting that water adsorption does not oxidize the defective
surface. However, ultraviolet photoemission spectroscopy ( UPS ) does distinguish
between them, suggesting that changes in the electronic energy levels at the vacancy
do occur [119] . Surface hydroxyls are known to be important in the photocatalytic
applications of TiO
2
, so Valentin and Pacchioni set out to investigate the electronic
Figure 8.22 Reaction scheme for the adsorption of water at
an O
2c
vacancy on TiO
2
(110). After adsorption there is the
possibility of electron reorganization to give OH radical
species.
8.4 Calculation of Surface Structure 377
nature of the surface hydroxyls and oxygen defect structures to attempt to clarify
their electronic structure [120] . Their work compares the gradient - corrected DFT
functional PBE and the hybrid B3LYP methods using the CRYSTAL localized basis
set code. In the calculations the full electronic structure, including core states, is
optimized self - consistently, avoiding the use of pseudopotentials.
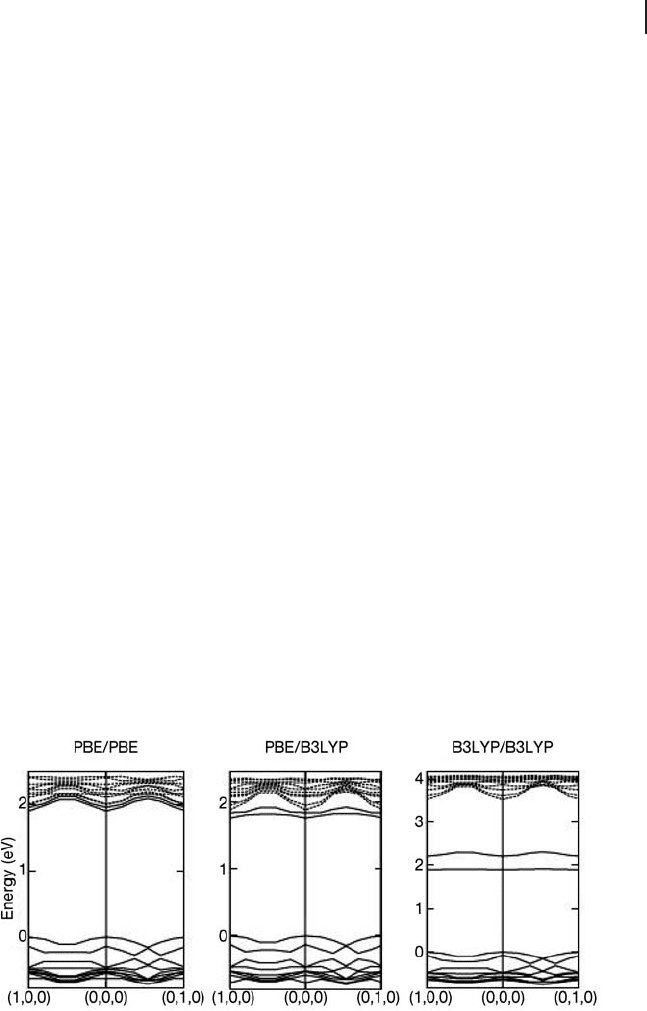
Figure 8.23 compares the calculated band structures from PBE optimized geom-
etries with B3LYP results both at the PBE geometry and after re - optimization of
the atomic co - ordinates using the hybrid functional. In the PBE/PBE result the
bands due to the hydroxylated Ti atom orbitals are contained within the conduction
band states, much as the surface states of the perfect surface. Density and spin
plots also show that the two electrons associated with the Ti
3+
centers in Figure
8.22 are spread over the Ti ions of the entire slab. The geometries obtained from
singlet and triplet electronic states are also identical and show only minor changes
of bond lengths around the defect site. The Fermi level in this PBE calculation lies
in the conduction band and so the pure functional is unable to reproduce the
electronic structure even qualitatively. Fixing the PBE calculated geometry and
carrying out a B3LYP calculation (PBE/B3LYP) does move the defect states out of
the conduction band but the separation is small. The B3LYP geometry optimiza-
tion gives different geometric structures for the singlet and triplet states, with the
triplet 0.6 eV lower in energy. This B3LYP/B3LYP calculation gives a larger band
gap than PBE as expected from the work of Zhang and coworkers on the
dependence of E
G
on the amount of exact exchange presented in Figure 8.6 . The
calculated adsorption energy for water at the vacancy site following the scheme
in Figure 8.22 is 1.8 eV, which is in reasonable agreement with the experimental
heat of adsorption of H
2
O to this surface estimated using temperature programmed
desorption (1.4 eV). The band structure from the B3LYP optimization shows two
bands around 1 eV below the bottom of the conduction band. These bands are
Figure 8.23 The calculated band structure for the O
2c
defective TiO
2
(110) surface with the geometry optimized
using PBE and bands calculated at the PBE (PBE/PBE) or
B3LYP (PBE/B3LYP) levels and with the geometry and bands
calculated using B3LYP (B3LYP/B3LYP). From ref. [120] .
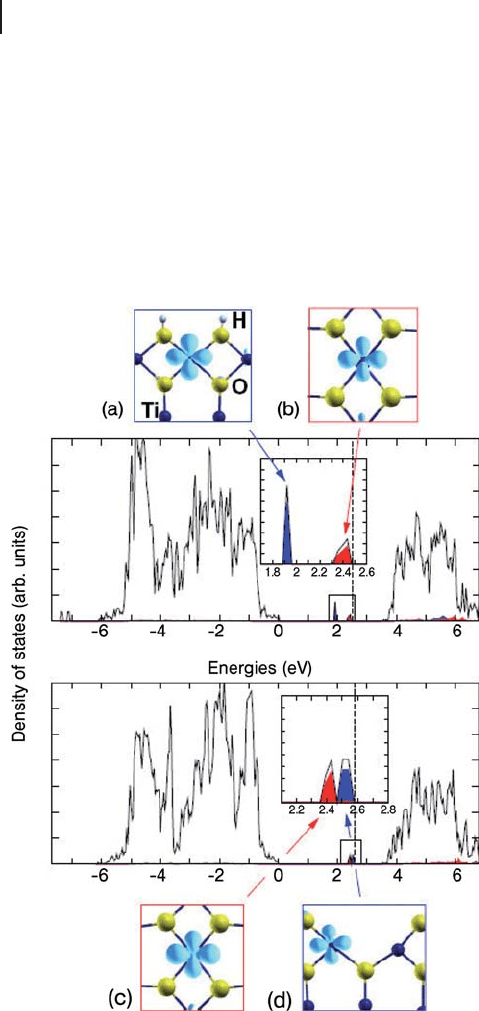
378 8 Theory: Periodic Electronic Structure Calculations
narrow, indicating electron localization, and closely associated with two particular
Ti surface ions. The fi rst is the Ti
6c
between the two hydroxyl groups, while the
second is a nearby Ti
5c
. The d orbitals involved are shown along with the calculated
density of states plot of Figure 8.24 . The lower of the two bands is for the Ti
c6
3+
ion
(Figure 8.24 a) while the singly occupied d orbital of the under coordinated Ti
5c
3+
ion gives a gap state nearer the conduction band (Figure 8.24 b). The geometry
relaxation of the surface leads to elongation of the Ti
3+
–
O bonds by up to 0.1 Å
owing to the charge localization at these metal centers.
Figure 8.24 Calculated density of states for the O
2c
defective
TiO
2
(110) surface (lower plot) and the same surface after
hydroxylation (upper plot). Only the majority spin DOS is
shown in each case. The metal orbitals responsible for the
gap states are picked out in images (a) – (d). From ref. [120] .
8.4 Calculation of Surface Structure 379
Calculations at the B3LYP level on the defective surface also show two localized
d orbital states in the band gap. These are associated with the Ti
5c
3+
site formed by
removal of the O
2c
atom (Figure 8.24 d) and a nearby Ti
5c
ion (Figure 8.24 c) with
the former higher in energy than the latter. This arrangement gives a smaller
repulsion between the two electrons left by the vacancy formation than would
placing them either side of the defect site as suggested in Figure 8.22 . Comparing
the calculated density of states in Figure 8.24 for the defective and hydroxylated
surface confi rms that there is a shift in the occupied energy levels, with the level
associated with the Ti coordinated by O
2c
oxygen atoms shifting down in energy
by around 0.6 eV on adsorption of water. However, both structures indicate that
the electrons left at the defect site by Equation 8.42 reside at metal centers, so that
water does not act as a oxidizing agent in this case.
As an example application of the DFT + U method to the study of anion vacan-
cies we turn to molybdenum oxide, MoO
3
. Oxides of molybdenum have been
known for some time to show activity for oxidation catalysis in which the oxide
surface itself acts as the oxygen donor in Mars – van Krevelen - based mechanisms
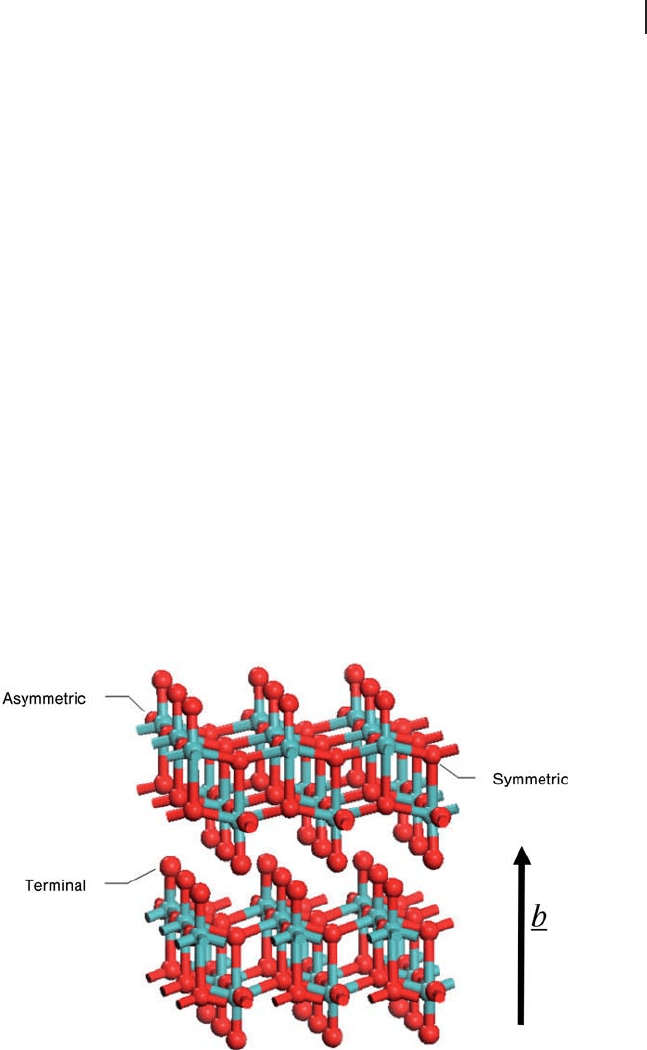
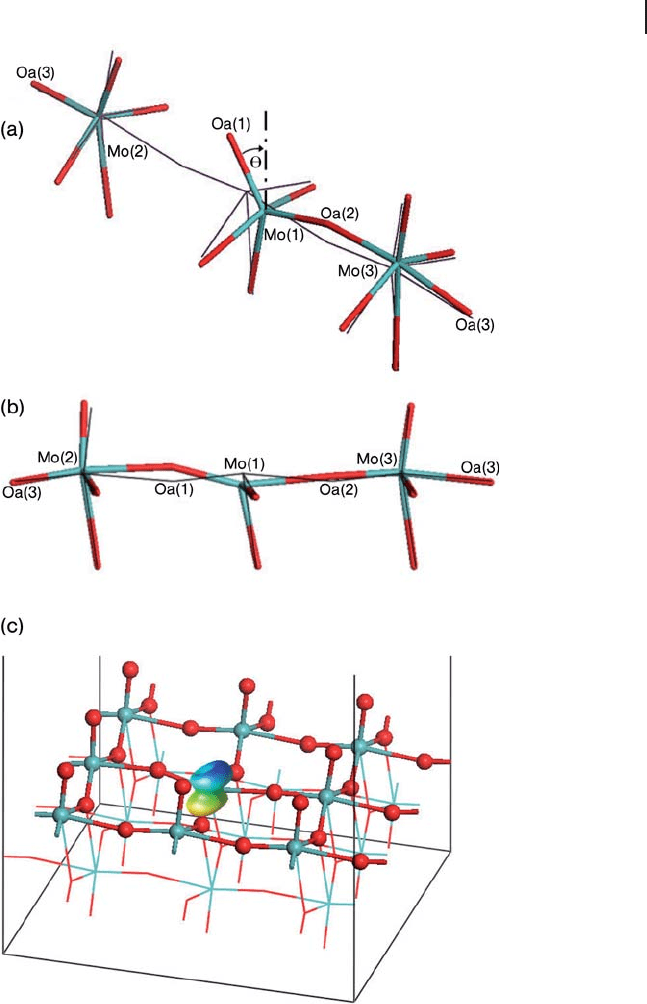
[121 – 123] . The structure of molybdenum trioxide is shown in Figure 8.25 and
consists of bi - layers formed from edge - sharing MoO
6
octahedra, although there is
considerable distortion away from O
h
symmetry at Mo. There are three structurally
distinct oxygen atoms: asymmetric, symmetric and terminal. The asymmetric
bridging oxygen atoms are twofold coordinate, forming one long (2.25 Å ) and one
short (1.73 Å ) bond with two Mo atoms in the same layer. The symmetric bridging
oxygen atoms are threefold coordinate, with two equal bonds (1.95 Å ) to Mo atoms
in the same layer and one much longer interaction (2.33 Å ) with a Mo atom of the
Figure 8.25 Two bilayers of bulk α - MoO
3
. The three different
types of O atoms are indicated (terminal, symmetric and
asymmetric). Oxygen, red; molybdenum, blue.

380 8 Theory: Periodic Electronic Structure Calculations
other sub - layer. Finally, the terminal oxygen is bonded to only one Mo atom,
forming the shortest Mo
–
O bond in the system (1.67 Å ). The crystal unit cell is
orthorhombic, with parameters a = 3.9628, b = 13.855 and c = 3 6964.Å
′
for a unit
cell containing Mo
4
O
12
[124] . The bi - layers are formed parallel to the (010) plane
with no chemical bonds between them, making this the easiest surface to
cleave.
MoO
3
is actually the fully oxidized end - member of a series of materials with
stoichiometries from MoO
2
to MoO
3
. For example, Wang and coworkers [125]
identifi ed two molybdenum sub - oxides Mo
18
O
52
and Mo
8
O
23
by means of electron
diffraction and high - resolution transmission electron microscopy ( HRTEM ) in
combination with image simulation. These are derived from MoO
3
by crystallo-
graphic shearing to accommodate the large number of oxygen vacancies. However,
the materials are related to the basic structure of MoO
3
with the Mo
18
O
52
(100)
surface built up of MoO
3
(010) terraces with MoO
3
(100) edges and MoO
3
(001) kinks
[126] . Since the unit cells of these types of materials are complex, modeling studies
have concentrated on the perfect MoO
3
(010) and its point defects as representative
of the terrace regions in the sub - stoichiometric material.
Different theoretical approaches have been used to study MoO
3
surfaces. Two
major ab initio periodic HF studies were published almost at the same time by
Papakondylis and Sautet [127] in 1996 and Cor à and coworkers [128] in 1997.
Papakondylis and Sautet used the CRYSTAL program to carry out HF - level calcula-
tions, with correlation energy estimates added using the PW91 functional with the
HF density. They showed how the structure can thought of as built up from an
MoO
3
molecule to the three - dimensional structure via a chain polymer and ribbon
based on the components of the crystallographic bi - layers. The evolution of the
band structure from the MoO
3
molecular orbitals could be followed through this
sequence. The (100) surface of MoO
3
contains penta - coordinated Mo atoms, which
are accessible Lewis acid centers. At the time automated geometry optimization
was not possible, but the interaction of adsorbed H
2
O with the fi xed surface were
optimized “ by hand ” based on the position and orientation of the molecule over
the surface. This showed that the adsorption of H
2
O molecules on these sites is
favorable but the electron transfer between the surface and adsorbate was actually
quite small.
Cor à and coworkers also used HF and the localized basis set approach of
CRYSTAL with a posteriori correlation corrections from PW91 [127] . They opti-
mized the bulk structure by systematically varying each degree of freedom inde-
pendently, starting with the weakest interaction fi rst. This is the interlayer spacing
for which they found a minimum, both at the correlated and the HF level. The
GGA method gave an underestimated interlayer spacing while HF gave an over-
estimate, consistent with the expected under - binding in GGA approaches. The fact
that minima are found at all indicates the presence of a weak attractive Coulombic
force between bi - layers since the long - range correlation responsible for van der
Waals interactions are absent in both approaches. They then analyzed the ground -
state electronic properties of MoO
3
, giving the electron density map shown in
Figure 8.26 a. Here the difference between the calculated density and that of a set
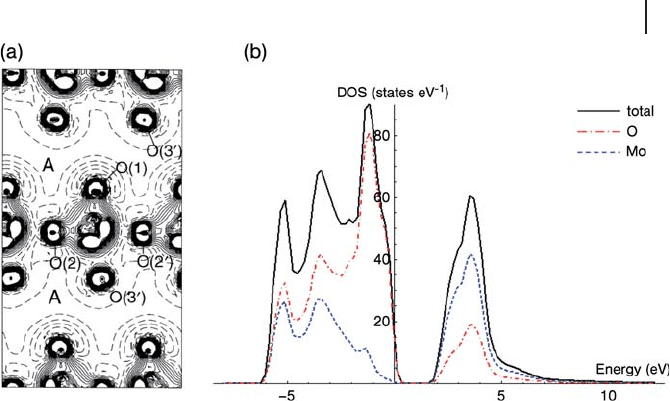
8.4 Calculation of Surface Structure 381
of non - interacting ions placed at the lattice positions is plotted to show where
charge has shifted to because of bonds formed between the ions. There is a net
loss of charge from the interlayer region and a particularly notable increase in
charge density between the Mo at the center of the fi gure and the associated ter-
minal O atom. This, along with a Mulliken charge analysis, shows that the nature
of the Mo
–
O interaction changes considerably according to the oxygen type, from
strongly covalent for the shortest bond (terminal oxygen) to a predominantly ionic
interaction for the longest bonds (interlayer Mo
–
O(symmetric)). The oxygen
atoms denoted O(2) and O(2 ′ ) in Figure 8.26 a are asymmetric, and this is clearly
refl ected in the asymmetry of the accumulation of charge density between the
central Mo and O(2) or O(2 ′ ). The DOS plot from our own work [129] using PW91
functionals in the VASP code is shown in Figure 8.26 b. The calculated valence
band region has a width of 5.8 eV. The computed band gap is 2.1 eV and, as
expected for a GGA method, this is an underestimate of the experimental value.
MoO
3
is an n - type semiconductor with an indirect band gap between 2.9 eV and
3.15 eV [130] . The DOS shows that the valence band contains both metal and
oxygen contributions, confi rming that there is a considerable covalent character
to the Mo
–
O bonds. Even so, the top of the valence band is largely O(2p) in
Figure 8.26 (a) Difference electronic charge
density map (calculated bulk minus isolated
Mo
6+
O
2 −
ion densities), displayed in the
crystallographic plane containing the central
Mo and the vertices O(1)O(2 ′ )O(3 ′ )O(2) of
one isolated MoO
6
octahedron. Continuous,
dashed and dot - dashed lines correspond
to positive, negative and zero difference
respectively. The interval between the
isodensity lines is 0.005 a.u. (electrons a
0
−3
).
The map extends beyond the central
octahedron and includes part of the interlayer
space, denoted by A. Taken from ref. [128] .
(b) Total DOS curve (black solid) with
decomposition into molybdenum (dashed)
and oxygen (dot - dashed) contributions, for
the MoO
3
bulk system. The energy origin is
chosen as the highest occupied state and a
Gaussian smearing width of 0.2 eV is applied.

382 8 Theory: Periodic Electronic Structure Calculations
character and the bottom of the conduction band consists largely of Mo orbitals
so the band gap is of a charge - transfer type.
Equation 8.42 indicates that the removal of a surface oxygen ion leaves two
electrons to be accommodated by the surface. For the terminal oxygen vacancy on
the MoO
3
(010) surface we have compared PBE and PBE + U calculations using
the VASP code [129] . In the PBE calculations the two electrons move into states
in the conduction band and are spread evenly between the Mo centers of the simu-
lation slab. In these calculations the surface structure is changed considerably,
with one of the asymmetric oxygen atoms neighboring the defect moving to
replace the lost terminal oxygen atom (Figure 8.27 a). In the PBE + U case the two
electrons are localized, reducing the single Mo ion from Mo
6+
to Mo
4+
and giving
a triplet ground state. This is confi rmed by a plot of the spin density difference
( ρ
α
( r ) − ρ
β
( r )) which shows the two electron spins localized at the metal center
where the terminal oxygen atom was removed (Figure 8.27 c). The corresponding
surface relaxation is considerably reduced, with the bridging symmetric and asym-
metric oxygen atoms staying in plane (Figure 8.27 b). As far as the rest of the lattice
is concerned, the electronic structure of the Mo
4+
center must closely resemble the
original Mo
6+
=
O species. The value of U in these calculations was set to a value
of 6.3 eV by comparing the calculated spin density with a reference cluster calcula-
tion at the B3LYP hybrid functional level.
Testing the accuracy of calculations on defects is more diffi cult, since experi-
mentally the defects are a minority and randomly distributed species. However,
we have been to make comparisons with surface spectroscopy results. Queeney
and Friend have synthesized oxidized Mo(110) surfaces with specifi c types of
oxygen coordination and characterize them using infrared and electron energy loss
techniques [131, 132] . During the oxidizing process, two distinct vibrational fre-
quencies υ (Mo
=
O) peaks were resolved at 992 and 1016 cm
− 1
using infrared spec-
troscopy. These are higher in frequency than any other surface vibrations owing
to the high bond order between Mo and terminal oxygen atoms. When total oxida-
tion was achieved by maintaining a fl ow of O
2
following high - temperature oxida-
tion, all terminal (Mo
=
O) sites were populated, and a single υ (Mo
=
O) at 996 cm
− 1
was observed. Using the numerical second derivative approach the vibrational
frequencies of M
=
O
term
were calculated for our perfect surface and for the terminal
oxygen groups neighboring the defect site. This gave values of 1023 cm
− 1
and
995 cm
− 1
suggesting that the high - frequency peak seen experimentally is in areas
that are relatively defect free while the lower frequency may be assigned to termi-
nal oxygen atoms near to defects in the partially oxidized fi lm.
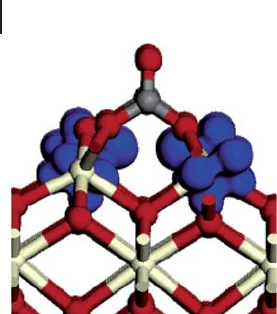
Our fi nal example is a study using DFT + U of the oxidation of CO over ceria.
This concerns the reduction of surface sites with the simultaneous oxidation of
an adsorbate. According to PW91 calculations [108] the most stable surfaces of
fully oxidized CeO
2
are (111), (110) and (100), with calculated surface energies
of 0.68, 1.01 and 1.41 J m
− 2
respectively. Nolan and Watson [133] have carried out
calculations on CO adsorption over these preferred surfaces using PW91 coupled
with the Hubbard model using a value of U = 5 eV based on studies of the
defective CeO
2
surface [134] . The adsorption of CO on the (111) surface is found
8.4 Calculation of Surface Structure 383
Figure 8.27 (a) Chain of sites along the asymmetric oxygen
direction taken from the PBE relaxed MoO
3
(010) surface
with a terminal oxygen atom vacancy; the structure of the
perfect surface is overlaid as a line drawing. (b) As (a) for the
PBE + U optimization. (c) The calculated spin density for the
PBE + U calculation showing the full simulation cell for parts
(a) and (b).
384 8 Theory: Periodic Electronic Structure Calculations
to be typical of physisorption, with an interaction energy of only − 25 kJ mol
− 1
and
no structural alteration of either CO or the surface. Over the other two planes,
initial placement of the CO molecule in a surface site bridging two oxygen ions
resulted in the spontaneous formation of a carbonate
CO
3
2−
species. This involves
removal of two O atoms and two electrons from the surface and so two Ce centers
are reduced from Ce
4+
to Ce
3+
at the same time. In the calculated spin densities,
Figure 8.28 , the two reduced Ce centers are clearly seen with the additional elec-
trons entering f states.
8.5
Conclusions
We have tried to highlight in this chapter the differences between the major
computational methods for the simulation of the electronic structure of materials
based on periodic boundary conditions. HF theory has a well defi ned theoretical
framework but ignores correlation energy from the outset. This is introduced
via the density in LSDA and GGA methods but at the expense of a less well
defi ned treatment of exchange. Even so the surface structure of stoichiometric main
group oxides is well described by either approach. The electronic states are less sat-
isfactory, with HF tending to severely overestimate and DFT to underestimate band
gaps. For catalysis, the empty states in the conduction band or in gap states are
responsible for Lewis acidity and so an accurate description of the energies of
these states is desirable. We have seen how hybrid functionals can correct for the
underestimation of the band gap in DFT by including some proportion of exact
exchange.
For transition metal oxides with partially fi lled d states, the balance between
Coulomb and exchange interactions is even more critical. DFT strongly underes-
timates the exchange gap present in materials with magnetic order such as NiO,
while it is again overestimated in HF. This can be corrected either through hybrid
functionals or with the DFT + U approach.
Figure 8.28 The spin density (blue) for CO
chemisorption on the (100) surface of ceria.
Taken from ref. [133] . Oxygen, red; Carbon,
grey; Cerium, light yellow.