Jackson S.D., Hargreaves J.S.J. Metal Oxide Catalysis
Подождите немного. Документ загружается.


8.4 Calculation of Surface Structure 365
of the water O atom (O
w
) with a low coordination Al ion through a dative bond in
which charge is donated from the adsorbate into the Lewis acid center. This leads
to a calculated binding energy of 97 kJ mol
− 1
which increases by a further 30 –
40 kJ mol
− 1
on heterolytic dissociation of an O
–
H bond to form two surface hydroxyl
groups. The range of energies stems from the choices possible for the surface
oxygen that receives the H
+
in the fi nal state.
The free energy of dissociation based on constrained dynamics also showed that
the calculated barrier depends on the geometry of the fi nal state. If an O neighbor
of the Al
3+
to which molecular oxygen was adsorbed is used to receive H
+
, the
transition state is a strained four - membered ring and a barrier of 28 kJ mol
− 1
is
obtained. This reduces to only 9 kJ mol
− 1
when the pathway to place H
+
on a second
neighbor oxygen is used. The second neighbor has the advantage that the transi-
tion state formed is a six - membered ring and so the O
w
–
H
–
O
surf.
angle is nearer
to linear at 166 ° , compared to 135 ° in the case of the nearest neighbor.
The geometry of the transition state has also been found to be important for the
dissociation of HF on α - Al
2
O
3
(0001) and the subsequent halogen exchange with
CH
2
Cl
2
based on DSOLID calculations [102] .
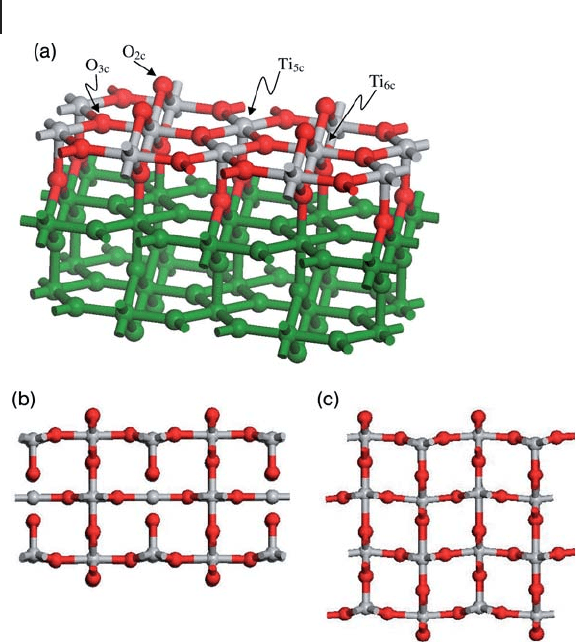
One of the easiest cleavage planes on rutile - structured TiO
2
corresponds to
the (110) surface. In the bulk structure [103] all Ti atoms are six coordinate with
four equivalent equatorial Ti
–
O bonds (1.97 Å (PBE)) slightly shorter than the
two axial (2.00 Å (PBE)). The preferred termination of the (110) surface is shown
in Figure 8.17 a and contains a mixture of sixfold (Ti
6c
) ions joined by surface
bridging oxygen atoms (O
2c
) and fi vefold (Ti
5c
) coordinate surface cations. Com-
pared to bulk centers the Ti
5c
atoms have lost one axial Ti
–
O bond. The line of
bridging oxygen atoms on the bulk termination is mirrored below the surface
plane and so a neutral stacking unit consists of an atomic tri - layer. For the simula-
tion of this surface the choice of the number of these tri - layers in the slab is
important. Figures 8.17 b and c show the result of relaxations using the SIESTA
code with three and four tri - layer slabs respectively. In both cases the Ti
5c
atoms
move down into the surface toward their remaining axial oxygen atom, but this is
a small effect compared with the surface ion movement seen for α - Al
2
O
3
(0001).
In addition, the four tri - layer slab is deformed and the surface appears to optimize
to a rumpled structure. In the thinner three tri - layer slab the surface is less dis-
torted from the bulk termination. Pacchioni and coworkers [104] have pointed out
that slabs with an odd number of tri - layers have a symmetry plane in the middle
of the slab that is absent for even numbers of stacking units. To investigate this
difference they used the PW91 functional, and compared results from the localized
basis set code CRYSTAL and plane - wave basis set code VASP. The calculated bulk
band gap from the plane - wave approach is around 0.2 eV lower than with the local-
ized basis set and this is ascribed to the use of ultra - soft pseudopotentials in the
VASP calculations. The different behavior for odd and even numbers of tri - layers
also results in oscillations in the band gap with slab thickness, which are mainly
due to changes in the calculated position of the bottom of the conduction band.
These states are associated with the coordinatively unsaturated Ti
5c
surface atoms
and are composed largely of the d orbitals with lobes perpendicular to the surface.
366 8 Theory: Periodic Electronic Structure Calculations
However, unlike the α - Al
2
O
3
(0001) example, these are not separate gap states at
the surface but simply the lowest lying states of a band that also contains mixed
Ti
–
O crystal orbitals.
In Pacchioni ’ s calculations it was found that the occupied bonding Ti
–
O hybrid-
ized orbitals tend to form bonds that are stronger between the surface tri - layer and
the fi rst sub - surface one than are found between tri - layers in the bulk. For the
even slabs this leads to a tendency to form a set of paired tri - layers, which give
rise to the observed rumpling, whereas for odd slabs a more regular spacing of
the tri - layers in the slab center is achieved.
The (110) surface of TiO
2
has also been subjected to LEED - IV experiments,
giving direct information on the surface structure [80] . Thompson and Lewis have
used the odd - numbered tri - layer slabs recommended by Pacchioni to consider the
Figure 8.17 (a) The TiO
2
(110) surface with surface Ti
6c
and
Ti
5c
atoms. Titanium, silver; oxygen, red; other atoms in the
slab, green. (b) The relaxed structure of the three tri - layer slab
viewed in cross - section. (c) The relaxed structure of the four
tri - layer slab. Atom colors for the surface layer of (a) are used
throughout (b) and (c).

8.4 Calculation of Surface Structure 367
accuracy of the surface geometry from calculations compared to this data [105] .
They employed the plane - wave basis set code VASP with the PW91 functional.
Their results, for the surface layer atoms, are reproduced in Table 8.6 and shows
that the surface relaxations are converged for slabs of nine tri - layers. This table
also includes results of earlier calculations by Gale and Harrison, who compared
LDA and HF methods for the same surface and who employed seven tri - layer slabs
[106] . Gale and Harrison found that the bridging O
2c
atoms remain at their posi-
tions in the simple bulk termination whereas the experimental data and the more
recent calculations indicate a signifi cant outward movement. However the metal
ion positions are better reproduced in the earlier work.
8.4.3
Infl uence of Environment on Surface Structure
It is clear from experiments that the character of an oxide surface is dependent
on its environment. For example, surfaces may be prone to hydroxylation and
many catalytic processes require reduction/oxidation cycles that must create/
replenish oxygen ion defect sites at the surface. If the surface composition of an
oxide is different to the bulk, the surface energy can no longer be defi ned as simply
the difference between the bulk and slab calculations per unit surface area.
However, the equilibrium surface energy can still be obtained from calculated
energies if we turn to the idea of the chemical potential [94] . The surface of an
oxide should be in equilibrium with both the oxide bulk and the atmosphere above
the oxide and so the chemical potential of all species must be constant in the three
regions, bulk, surface and atmosphere. The simplest case to consider is the effect
of oxygen partial pressure on the surface stoichiometry. For an oxide of formula
A
m
O
n
the formation of a non - stoichiometric surface in a slab simulation can be
thought of as the chemical reaction
N
m
N
mn N n
m
NN
A
eO
AO O A O
A
AeO
+→
+
(8.32)
Table 8.6 The displacement of surface atoms from their bulk positions in TiO
2
(110).
Expt [79] Method (Number of tri - layers in slab)
PW91 (5)
[105]
PW91 (7)
[105]
PW91 (9)
[105]
PW91 (11)
[105]
LDA (7)
[106]
HF (7)
[106]
Ti
6c
0.25 ± 0.03
0.29 0.33 0.42 0.43 0.22 0.25
Ti
5c
− 0.19 ± 0.03 − 0.10 − 0.07 − 0.03 − 0.03 − 0.17 − 0.17
O
2c
0.25 ± 0.03
0.09 0.13 0.23 0.23 0.01
− 0.01
O
3c
0.27 ± 0.08
0.24 0.27 0.32 0.32 0.13 0.11
368 8 Theory: Periodic Electronic Structure Calculations
where the formula on the right hand side is that of a simulation slab and the
additional term on the left, required to balance the equation, can be thought of as
coming from gas - phase oxygen. The term N
eO
defi nes the oxygen excess in the
slab and can be negative.
The surface free energy, γ , is just the free energy for the reaction of Equation
8.32 per unit surface area of the slab. If a slab simulation geometry gives two sur-
faces of area S :
γµ=
1
2
−−
()
S
G
N
m
gN
slab
A
AO eO O
(8.33)
where G
slab
is the free energy of the slab representation of the surface and g
AO
is
the free energy per stoichiometric unit of the bulk material. The chemical poten-
tial, µ
O
, is used to obtain the free energy of the excess O species. It is useful to
defi ne the oxygen excess per unit surface area, Γ
O
, when Equation 8.33 becomes:
γµ=
1
2
−
()
−
S
G
N
m
g
slab
A
AO O O
Γ
(8.34)
To link this defi nition of surface energy to calculations on relaxed slabs we now
make the assumption that the entropy in the slab and bulk oxide free energy
terms can be neglected. Then the surface energy based on calculated quantities
gives
γµ=
1
2
−
()
−
S
E
N
m
e
slab
A
AO O O
Γ
(8.35)
in which E
slab
is the energy of the geometry optimized slab defi ned by the right
hand side of Equation 8.32 and e
AO
is the energy per formula unit of a relaxation
of the bulk. Although Equation 8.35 defi nes the surface energy, it still requires the
chemical potentials for the excess oxygen to be set. The oxygen chemical potential
under any given conditions of temperature T and partial pressure P
O
2
can be
estimated using:
µµ
OO
0
O
=+
(
)
1
2
2
0
RT
P
P
ln
(8.36)
where the superscript 0 is used to indicate standard conditions and R is the molar
gas constant. Usually the zero of chemical potential would be taken as
the value for dioxygen gas under such standard conditions. However, using
calculated data the energy is referenced to a state in which all atom cores and
valence electrons are isolated and so an estimate of the energy of the dioxygen
molecule is required to continue the calculation. The assumptions used by DFT
and other fi rst principles methods that can be applied to the solid state mean that

8.4 Calculation of Surface Structure 369
the accuracy of the calculation of such absolute values of molecular energy is not
great. It is better to estimate the standard state chemical potential using a cycle
which gives µ
O
0
as the difference between more reliable quantities such as lattice
energies.
For example the standard energy of formation of the oxide is the energy for the
reaction:
m
n
As O g A O s
2mn
()
+
()
→
()
2
(8.37)
for which,
∆Ggmn
mnf
0
AO
0
A
0
O
0
AO
()
=− −µµ
(8.38)
Since the metal will usually have a standard state which is solid the values
g
AO
0
and
µ
A
0
can be obtained from extrapolation of DFT optimization calculations.
Accurate values of ∆G
mnf
0
AO
()
are tabulated for the common oxides [56] , for
example the value for Al
2
O
3
is − 1576 kJ mol
− 1
and so Equation 8.38 provides a route
to
µ
A
0
without explicit calculations on O
2
.
Equation 8.38 also sets the lower limit of the range of values accessible for µ
O
since reduction of the oxide will occur spontaneously into the pure metal and
dioxygen gas if the free energy for Equation 8.37 is positive. This means for a
stable oxide:
µ
O
AO A
>
−gmg
n
mn
(8.39)
An upper bound for µ
O
can be estimated from the point at which O
2
(g) begins
to condense on the surface. The thermodynamically stable composition of the
surface can now be predicted over the range of accessible partial pressures of O
2
by using the pressure dependence of the chemical potential from Equation 8.36
in Equation 8.35 .
This approach was fi rst applied to α - Al
2
O
3
(0001) by Finnis and coworkers [107]
who demonstrated that the Al termination which forms the type II non - polar
surface is stable under normal conditions. Elevated oxygen partial pressures
(several tens of atmospheres) would be required to make an oxygen - terminated
surface the preferred form. Reduction of the surface will not be favorable until the
lower limit of oxygen partial pressure is met, at which point bulk reduction is
favorable anyhow.
These thermodynamic arguments can also consider the hydroxylation of sur-
faces by including the chemical potential of hydrogen and/or water [108] . This type
of analysis shows that Al
2
O
3
surfaces of the α - phase are either Al or OH terminated
under all accessible conditions. However, the infl uence of the environment is
different for each Miller index owing to variation in the calculated surface energies.

370 8 Theory: Periodic Electronic Structure Calculations
Phase diagrams specifi c to each experimentally observed face have been derived,
which show that conditions should exist for which the (0001) surface is dehydroxyl-
ated, to give an Al termination, while other surfaces remain fully hydroxylated.
For transition metal oxides the surface composition can be altered more readily
by the partial pressure of oxygen. For example, RuO
2
(110) has been shown to move
from an over - oxidized state for high O chemical potential to a reduced form at the
lower end of its accessible range [109] . In such cases the electrostatic arguments
against “ polar ” surfaces have to be adapted in the light of surface cation reduction.
It should also be remembered that these calculations give the thermodynamic
stability of a given surface composition and do not take account of any kinetic
barriers to the required changes.
8.4.4
Transition Metal Oxides with Partially Filled d Bands
The band picture of electronic states in solids works well when there is strong
overlap between neighbors in the lattice. This leads to broad bands and delocalized
electronic states. The early successes of band theory were in the description of the
physical properties of metals such as electrical and thermal conductivity [110] .
However, in transition metal oxides band structure alone is often not suffi cient.
Consider a d
8
system such as Ni
2+
in NiO. The usual picture would be to draw
localized orbitals at each Ni site which are split into three t
2g
and two e
g
energy
levels by the crystal fi eld. The eight d electrons per ion would then fi ll the energy
levels as shown on the right in Figure 8.18 . This is a highly localized picture of
the electronic structure since the sets of d electrons are associated strongly with
particular nuclei. If movement of electrons occurs between centers we have to
remove an electron from one of the d
8
centers, leaving a d
7
, and shift it to a neigh-
bor which becomes d
9
. The electron – electron repulsion between the d electrons
will be proportional to the number of pairs of electrons present at each center.
With two d
8
ions we have 56 electron pairs, while the d
7
+ d
9
in isolation have a
total of 57 electron pairs. So the movement of electrons between centers requires
energy, effectively creating an energy gap. This type of argument was developed
by Mott and Hubbard to explain the insulating properties of transition metal
oxides with partially fi lled d shells. They introduced a parameter, U , which is used
to represent the Coulomb term as a function of d - orbital occupation so that the
relative energies of alternative arrangements can be estimated.
Figure 8.18 The localized electron populations of the t
2g
and E
g
states for two Ni
2+
d
8
ions in NiO. The average
confi guration is d
8
but transport of electrons through the
material would produce d
7
and d
9
centers.
8.4 Calculation of Surface Structure 371
A purely band structure - based treatment of this problem results in very narrow
bands for the d orbitals because of the weak overlap between neighboring Ni
2+
centers. However the band would be partially fi lled and so it would be concluded
that the system is metallic. This is the result generated by a simple LSDA approach,
as shown early on by Terakura and coworkers [111] . In the Mott – Hubbard analysis,
the localized picture becomes important when U is greater than the band width
predicted by band theory. In addition the electron confi guration affects the
exchange energy. For example, the two electrons in the triplet d
8
confi guration will
also have an exchange interaction and this is lost if one spin is reversed to produce
a singlet. This means that the e
g
energy levels appear lower in the triplet state than
the singlet so that an “ exchange gap ” can be introduced.
For transition metal oxides such as NiO the Mott – Hubbard model gives much
better agreement with experiment; localization of the electrons in this way explains
the anti - ferromagnetic ordering observed in NiO and its electrical insulator proper-
ties [112] . The terms required to calculate the electron – electron interaction energy
on a site, E
MH
UHF
, can be written in terms of the d - electron occupations, using a UHF
approach:
E
U
nn
UJ
nn
mm
mm
mm
mm
MH
UHF
=+
−
()
′
−
′
′
≠
′
∑∑
22
,,
,,
,,
,
ωω
ω
ωω
ω
(8.40)
where the sum over ω is over the spin states α and β , with − ω signifying the
opposite spin state to ω . The sum over m and m ′ considers each of the d states at
the site. In this way the fi rst term takes into account the Coulomb interaction
between electrons of opposite spin while the second term is for Coulomb interac-
tions between electrons of the same spin, with a correction for the exchange
interaction (rather confusingly given the symbol J ).
Of course some of these interaction terms are already present in an LSDA cal-
culation and so to incorporate this into a general DFT scheme requires these
duplicate contributions to be removed. One way to do this has been proposed by
Dudarev and coworkers [113] . They have derived a functional incorporating the U
and J parameters of the Mott – Hubbard model which is written:
EE
UJ
nn
mm
m
LSDA U LSDA U++
=+
−
()
−
()
∑
2
2
,,
,
ωω
ω
(8.41)
The advantage of this expression is that U and J appear only as their difference
( U − J ) and so it requires only a single parameter to be determined for any particu-
lar transition metal oxide under consideration. The J parameter shows little
system - to - system variability and is taken to be around 1 eV in most cases. Based
on a comparison of the calculated DOS and experimental electron energy loss
spectra ( EELS ) the U parameter for NiO was set at 6.2 eV. This gave a ground state
in which the Ni spins are aligned within (111) planes and neighboring planes have
372 8 Theory: Periodic Electronic Structure Calculations
opposite spin as shown in Figure 8.19 . This anti - ferromagnetic ordering and the
calculated site magnetization are in good agreement with experimental data. On
a practical level, the ordering of spins in this way means that the crystallographic
fcc unit cell cannot be used for the simulation as it contains an odd number of
(111) planes, and a supercell has to be employed which is commensurate with the
spin system. The calculated density difference between the LSDA + U and LSDA
calculations is plotted in Figure 8.20 . This shows that the inclusion of the Hubbard
model leads to a reduction of density in the interatomic region between Ni
2+
and
O
2 −
ions and increased localization of charges at the metal centers. The LSDA cal-
culation therefore overemphasizes the importance of covalent bonding between
the ions compared to the LSDA + U results.
More recently Rohrbach and coworkers have used NiO as an example to test
the implementation of the Hubbard model in the VASP code [114] . Their work
allows the combination of the PW91 gradient corrected functional with the on - site
Coulomb and exchange corrections given in Equation 8.41 . To set the U parameter
Rohrbach and coworkers plotted the bulk lattice parameter, metal center magnetic
moment and band gap over the range U = 1 to 9 eV. The best compromise for
accurate agreement with experiment on these three properties was U = 6.3 eV,
practically the same value as used in the earlier calculations of Dudarev and
coworkers [113] . The effect of the U term can be quantifi ed by comparing results
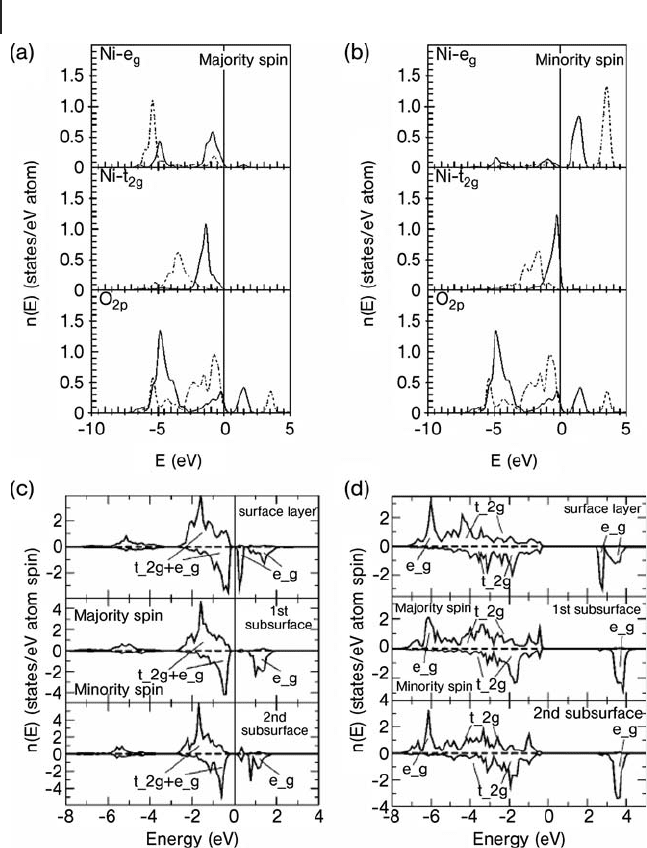
of calculations based on PW91 alone with those at this optimal value of U. Figures
8.21 a and b show an overlay of the DOS from unrestricted calculations for the
majority and minority spin states, respectively. At the PW91 level the band gap is
underestimated at 0.5 eV compared to the experimental values of between 4.0 and
4.3 eV. The calculations also indicate a simple Mott – Hubbard band gap with the
top of the valence band consisting of metal majority spin states of e
g
symmetry
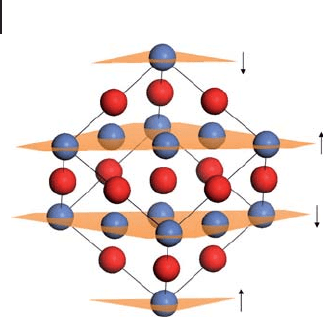
Figure 8.19 The antiferromagnetic AF
2
state of NiO. The (111)
planes through Ni
2+
centers are shown with arrows indicating
the orientation of the majority spin for each plane. Note that
the atoms at the top and bottom corners of the cell are
crystallographically, but not magnetically, equivalent. Nickel,
blue; oxygen, red.
8.4 Calculation of Surface Structure 373
along with minority t
2g
states and the bottom of the conduction band being just
minority spin e
g
states. The O(2p) contribution at these energies is relatively minor.
This means that the energy gap is created by the difference in energy levels for
different spin states. The majority degenerate e
g
states are occupied by two elec-
trons and so have an exchange contribution to their energy whereas the minority
e
g
states are empty. Thus it is an “ exchange splitting ” of the e
g
states that creates
the gap, which is the classic Mott – Hubbard argument for insulating behavior in
this type of oxide. In the PW91 + U this exchange splitting of the metal d orbitals
is increased to around 9.5 eV, because the electron localization increases the sta-
bilizing effect of the exchange interaction on the occupied majority e
g
states. Now
the top of the valence band has a largely O(2p) character, with contributions from
both majority and minority Ni t
2g
states. The lowest lying conduction band states
are mainly from the minority spin e
g
states, suggesting that the band gap is now
of mixed charge - transfer and Mott – Hubbard type. The band gap has also widened
to 3.2 eV, in much closer agreement with the experimental measurements.
Rohrbach and coworkers went on to consider the NiO(100) surface. Since NiO
is isostructural with MgO, this is a non - polar fl at surface corresponding to one of
the faces of the cubic unit cell. Reference to Figure 8.19 shows that the surface
will also have an anti - ferromagnetic structure with rows of surface cations having
parallel spins along the lines in which the (111) planes intersect the surface.
Relaxation with PW91 + U shows a very small (1%) inward movement of the
surface layer, but no buckling of the type observed for MgO(100) is reported.
Buckling only occurs at the LSDA level and has not been observed experimentally.
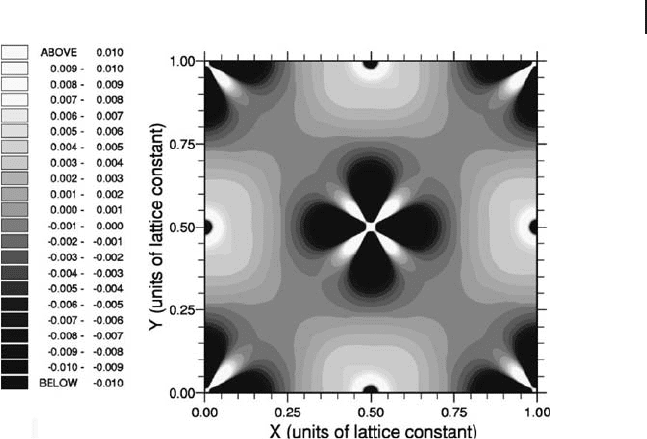
Figure 8.20 Charge density difference [LSDA + U ] − LSDA
(U = 6.2 eV) for bulk NiO taken through a (100) plane of the
rock salt cubic unit cell, centered on a Ni
2+
ion. Taken from
ref [113] .
374 8 Theory: Periodic Electronic Structure Calculations
Figure 8.21 Calculated densities of states for NiO, (a) bulk
structure at PW91 (solid lines) and PW91 + U (dashed lines)
for the majority spin, (b) as (a) for the minority spin.
(c) Breakdown of contributions to DOS by atomic layer for the
PW91 calculation and (d) breakdown by layer for PW91 + U .
In each case energies are referenced to the top of the valence
band. Taken from ref. [114] .