Jackson S.D., Hargreaves J.S.J. Metal Oxide Catalysis
Подождите немного. Документ загружается.


calculated using a gradient - corrected DFT technique so that the energy quoted
corresponds to the total energy of the valence electrons per unit cell. The bulk
MoO
3
lattice parameters are a = 3.963 Å , b = 13.855 Å and c = 3.696 Å [29] , so that
the
b
lattice vector is signifi cantly longer than
a
or
c
. This means that orbital
overlap is expected to be weaker in the b direction than in a or c and correspond-
ingly the dispersion in the band moving along
b
* is less. Hence the number of
grid points required in the
b
* direction is fewer than in
a
* or
c
* . From Table 8.1
we can see that the energy of the bulk is converged to a precision of 10
− 3
eV (or
0.1 kJ mol
− 1
) for a
k
- point grid of 5 × 3 × 5 .
This idea that large unit cell dimensions in real space require fewer samples to
be taken in reciprocal space can also be useful when working with supercells. A
supercell calculation uses multiple crystallographic cells as the repeated simula-
tion unit. For example, in calculations on surfaces this allows reconstruction on a
length scale longer than is allowed by the minimal unit cell. The sampling of
reciprocal space in supercell calculations can be reduced because multiple
k
- points
of the normal unit cell can be represented in real space. This idea is illustrated in
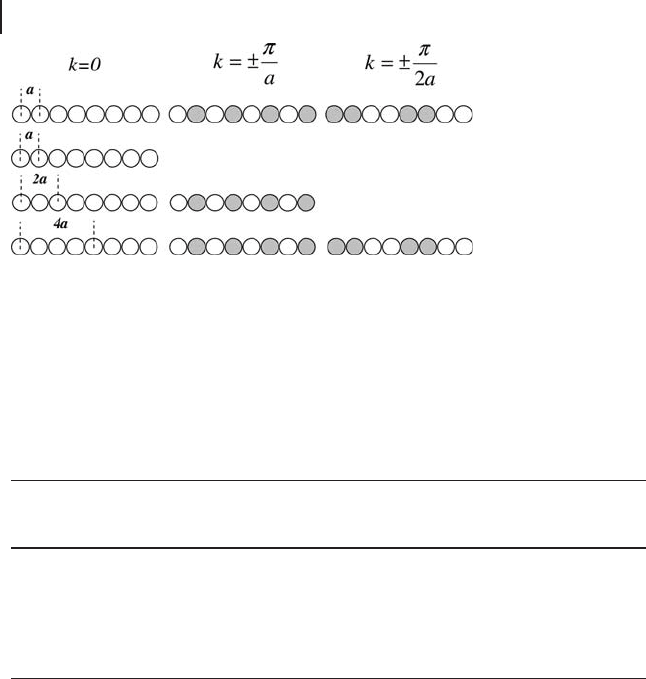
Figure 8.2 using a one - dimensional example with a single s - orbital per site. In the
minimal unit cell sampling at k = ± π / a corresponds to a wavefunction with phase
alternation between neighboring sites (in one dimension a * = 2 π / a ), which is at
the top of the band. If we use a supercell with a length of 2 a this wavefunction
can be represented with k = 0 because there are now two possible cell orbitals. The
total number of orbitals has not changed, but in the new cell one band would be
formed from the state with neighbors in the same phase and a different band
would be formed with neighboring states out of phase. The third state shown at
the top of the diagram would still require a reciprocal space point away from k =
0, as its repeat length is longer than 2 a. Doubling the cell once more allows all
three states to be represented in real space.
Table 8.1 Convergence of the bulk energy for MoO
3
, with respect
to the k - point grid using a planewave basis ( E
cut
= 520 eV) and
PBE functional within the VASP code.
k - point grid
a)
MoO
3
bulk energy (eV)
∆E
b)
(eV)
3 × 1 × 3 − 130.168 980
–
3 × 3 × 3 − 130.171 210
0.002 230
5 × 3 × 5 − 130.271 510
0.100 300
7 × 3 × 7 − 130.272 620
0.001 110
9 × 3 × 9 − 130.272 710
0.000 090
11 × 3 × 1 1 − 130.272 680
0.000 030
15 × 5 × 1 5 − 130.272 680
0.000 000
a Grid quoted as number of points sampled in the
a
* ×
b
* ×
c
*
reciprocal lattice directions.
b ∆ E is the difference in energy between two consecutive k - point
grids.
8.2 Electronic Structure Methods 335
336 8 Theory: Periodic Electronic Structure Calculations
Figure 8.2 One - dimensional example of using supercells
to reduce k - point sampling. With a minimal unit cell of
dimension a , the states shown on the top line require 3 k -
points to be sampled. Doubling the unit cell allows the fi rst
two states to be generated at k = 0; with a unit cell dimension
of 4 a all three states are contained within the local cell
wavefunctions.
This effect can be seen in calculations on real materials; Table 8.2 shows the
convergence of the binding energy per unit volume of α - Al
2
O
3
with cell size cal-
culated using gradient - corrected DFT with a localized basis set in the DSOLID
package [30] . The binding energy is with respect to DFT calculations on the neutral
atomic species and so the values quoted are considerably smaller in magnitude
than for the earlier MoO
3
example.
8.2.3.2 Basis Sets
A basis set is required to describe the cell periodic part of the Bloch function in
Equation 8.17 . It is possible to use localized functions based around the atomic
Table 8.2 The affect of unit cell size on calculated binding energy for α - alumina.
Multipliers for
a, b ( Å )
Formula of
repeating unit
Cell volume relative
to Rhomb. cell
Binding energy
c)
per
unit volume (eV Å
− 3
)
Rhomb.
a)
A l
4
O
6
1
− 0.6951
1,1
b)
A l
12
O
18
3
− 0.7175
2,1
b)
A l
24
O
36
6
− 0.7151
3,1
b)
A l
36
O
54
9
− 0.7173
4,1
b)
A l
48
O
72
1 2
− 0.7173
a The rhombohedral unit cell is the primitive cell; all other calculations employ the more usual
hexagonal unit cell.
b These structures were based on the experimental hexagonal unit cell with a = b = 4.759 Å ,
c = 12.991 Å and γ = 120 ° .
c Binding energy is defi ned as the difference between the total energy of the unit cell and the sum
of total energies for the same neutral atoms in isolation. This is quoted per unit volume to allow
comparison of the differently sized cells.

orbital like the basis sets found in molecular quantum chemistry. This is the
approach used in the CRYSTAL [31, 32] code which employs Gaussian functions
for the radial part of the atomic orbitals. The codes SIESTA [33] , DMOL
3
(and its
predecessor DSOLID) [34] also use localized basis sets but store the radial decay
of the basis functions on a numerical grid. For the localized basis set, work has
also to be undertaken to optimize the functions used for the solid state, as the
radial decay profi les used in molecular problems are usually not appropriate [35] .
For example, the O
2 −
ion is more diffuse than an oxygen atom in a molecule and
so the basis sets used for molecular quantum chemistry will not perform well. An
alternative approach is to use the periodic nature of the system to build the elec-
tronic wavefunctions as sets of plane - waves. Codes that take this avenue include
VASP [36] , DACAPO [37] and CASTEP [38] . The Carr Parinello molecular dynam-
ics code ( CPMD ) [39] is also a plane - wave code with the added feature that the
electronic degrees of freedom are included in the evolution of the system based
on Newton ’ s laws. The plane - wave basis is specifi c to the solid state and so some
background is given in the following text.
Any function used as the local part of the wavefunction description must
be periodic in the cell dimensions. In a plane - wave basis this is ensured by
choosing a linear combination of plane - waves with particular reciprocal space
vectors:
χ
jjG
G
cirGr
()
=⋅
()
∑
,
exp
(8.20)
in which the coeffi cient c
jG
is the amount of the plane - wave with reciprocal wave
vector
G
to include in local function j. Each
G
vector is described by the
equations
GT T a b c⋅= = + +2πmuvw
(8.21)
that is, the vector dot product between an acceptable
G
vector and any real space
vector,
T
, which is a combination of real space lattice vectors ( u,v,w are integers),
gives an integral number of 2 π radians. Under this condition any two points in
the lattice that are separated by a vector such as
T
must have the same value of
χ
j
(
r
). Note that the
G
vectors have a repeat which is contained within the unit cell
while the
k
- vectors used in the Bloch states of Equation 8.17 have periods on a
larger scale.
The use of plane - waves as a basis in this way is just like taking the Fourier
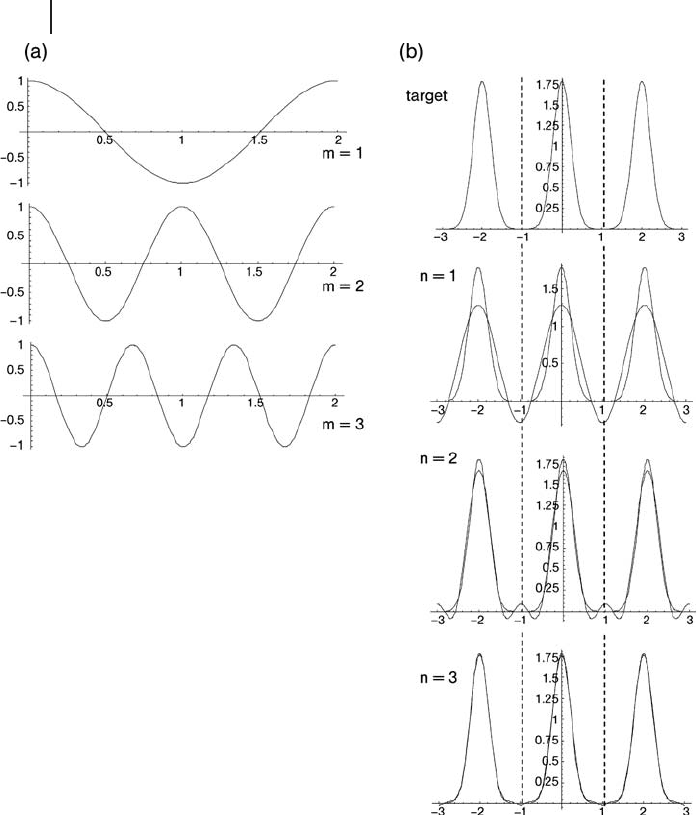
transform of a known function. A simple one - dimensional illustration of this is
shown in Figure 8.3 . Three waves which obey the periodic boundary condition
set by a unit cell of length 2 units ( m = 1 – 3 in Equation 8.21 ) are shown in Figure
8.3 a. The target function is a periodic set of Gaussians shown at the top of Figure
8.3 b. Since the target function is known, the coeffi cients, c
j
,
G
, can be calculated
analytically and the basis summed following Equation 8.20 . Figure 8.3 b shows
8.2 Electronic Structure Methods 337
338 8 Theory: Periodic Electronic Structure Calculations
Figure 8.3 (a) One - dimensional example of a plane wave
basis set for a unit cell of length 2 consisting of 3 functions.
(b) Application of the basis to generate a target function
which is a periodic set of Gaussians.
how the agreement between the target function and that constructed from the
plane - wave basis becomes better as the number of plane - waves used, n , is increased.
This Gaussian example is a particularly simple one because the function varies
quite smoothly with position, as do the plane - waves, and so after three basis func-
tions are included the match with the target is already very good. In a DFT calcula-
tion the target functions are not known and so the coeffi cients, c
j
,
G
, are solved for
as part of the self - consistent solution of the Kohn – Sham equations.
The plane - wave basis has the advantage that it can be systematically improved
by simply increasing the number of plane - waves used. The basis set is usually
truncated, based on an energy cut off, E
cut
, which defi nes the kinetic energy limit
for the plane - waves. The corresponding maximum
G
vector is related to the energy
cut off via,
G
max
=
2
2
π
h
mE
ecut
(8.22)
Here we have reverted to SI units so that m
e
(the electron mass) and h (the
Planck constant) appear in the formula. In most programs this |
G
|
max
value is used
along with Equation 8.21 to determine the number of plane - waves in the basis set
for the entire calculation by setting the maximum value of the integer, m. It has
been pointed out by Pulay that Equation 8.21 implies that the basis set is actually
dependent on the size of the unit cell, since
T
depends on the lattice vectors. This
can lead to additional stresses in calculations in which the cell volume is allowed
to vary. These appear because the quality of the basis set changes with cell dimen-
sions. In particular, when the volume of a unit cell is estimated using a plane - wave
basis, separate optimizations of the ions within a series of fi xed volume cells is
more reliable than allowing the cell volume to be optimized during a single run.
The goal of periodic calculations is to generate a reliable estimate for the electron
density of the unit cell. However, many chemical concepts of bonding rely on ideas
that are not so easily defi ned, such as atomic charges with which to quantify the
ionicity of the system. The expression for the electron density in terms of molecu-
lar orbitals Equation 8.16 does allow a partitioning of the charge density to be
undertaken to access such quantities. In terms of the basis set the density can be
re - written:
ρχχrr c r c rr
ji i
i
M
jk k
k
M
j
N
()
=
() ()
∑∑∑
=
dd
33
1
2
2
,,
*
(8.23)
where we have M functions in the basis set and c
j
,
i
is the coeffi cient for the expan-
sion of the i
th
basis function in the j
th
molecular orbital. Therefore the density is
actually built up from products of basis functions known as overlap functions. In
a localized basis set it is easy to assign basis functions to atoms, since each is based
on an atomic orbital belonging to a particular center. The basis functions belong-
ing to a particular atom, A , will appear in Equation 8.23 in some terms that involve
only functions centered on A and in some giving overlap with other centers. The
fi rst set of terms are usually assigned as belonging wholly to A , but to assign a
particular portion of the density to atom A we must also take some component of
the overlap with other atoms. Mulliken [40] proposed that these overlaps between
different atomic centers be divided equally between the atoms involved, and this
estimate of the atomic charge is widely reported in the output of periodic electronic
structure codes. When a plane - wave basis set is used in the calculation the assign-
8.2 Electronic Structure Methods 339

340 8 Theory: Periodic Electronic Structure Calculations
ment of basis functions to particular atoms is not so straightforward. A way around
this is to use the plane - wave basis to do the calculation and then fi t a local basis
representation to the resulting density and use the Mulliken analysis on that. The
fi tting process is often referred to as the projection of the plane - wave calculated
density onto the localized basis set.
Mulliken analysis is a quick and relatively simple way to break down the charge
density into atomic contributions but it is dependent on the basis set used. In
particular, if the number of basis functions is unevenly distributed between atoms,
those with rich basis sets will tend to have too much charge assigned through the
Mulliken procedure. To attempt to rectify this, methods based on the charge
density alone have also been developed. For example, Bader analysis uses the
minima in the density to defi ne a region around each atom over which the density
can be integrated numerically [41] .
8.2.3.3 Pseudopotentials
To describe the bonding between atoms, it is convenient to split the electrons for
each atom into valence and core states. In general, the core states are those not
directly involved in bonding interactions and the valence states are the outermost
atomic orbitals that can mix or otherwise interact strongly with neighboring atoms.
It is usual to replace the infl uence of the core electrons of an atom on the valence
states by the use of pseudopotentials, that is, a function which represents the true
full electron potential in the valence region but varies more smoothly in the core
region of the atoms. In the valence region, the wave functions from calculations
using pseudopotentials and those with all electrons included should match closely.
In the core region, the smooth variation of the pseudopotential leads to a more
smoothly varying wavefunction. In essence, the radial nodes in the core region are
eliminated. To be resolved correctly, rapid spatial variation of core state nodes
would require a large number of plane - waves in the basis set; hence the use of
pseudopotentials reduces the number of plane - waves that have to be used in a
calculation while maintaining a good representation of the chemically important
valence states. An additional benefi t for heavy elements is that relativistic effects
are more pronounced in the core region but their effect on the electrons in the
valence region can be reproduced by calibrating the pseudopotential against full
relativistic calculations on atoms. The valence electronic structure is then free to
use non - relativistic DFT.
Each pseudopotential is defi ned within a cut - off radius from the atom center.
At the cut - off, the potential and wavefunctions of the core region must join
smoothly to the all - electron - like valence states. Early functional forms for pseudo-
potentials also enforced the norm - conserving condition so that the integral of the
charge density below the cut - off equals that of the all - electron calculation [42, 43] .
However, smoother, and so computationally cheaper, functions can be defi ned if
this condition is relaxed. This idea leads to the so called soft and ultra - soft pseu-
dopotentials defi ned by Vanderbilt [44] and others. The link between the pseudo
and real potentials was formalized more clearly by Bl ö chl [45] and the resulting

projector augmented - wave pseudopotentials have also been implemented in plane -
wave basis set codes [46] .
In oxides, the division between core and valence states should be carried out
with care. For example, in strongly ionic materials the cation may have lost all
valence electrons to the anion and so the cation/anion interaction involves orbitals
on the cation that are atomic “ core ” states. In this situation, explicit inclusion of
the outermost cation orbitals would be required for accurate results. Even so, the
replacement of the core region with a smoother potential can reduce the calcula-
tion time even for H atoms, and so pseudopotentials are available even in this
case.
For consistency, pseudopotentials should be based on the same functionals as
used for the valence states. This point has been illustrated by Gale and coworkers
in their study of aluminum trihydroxides [47] . Table 8.3 compares their results of
cell optimizations using various mixtures of psuedopotential and valence state
functionals with the experimental structure of gibbsite [48] . LDA pseudopotentials
in an LDA optimization of the cell gives underestimated cell parameters, consis-
tent with the usual expectation that LDA gives over - binding in chemical bonds.
This is partially corrected by switching just the valence states to the GGA func-
tional PBE, which is a fortuitous consequence of over - binding in LDA being
compensated for by under - binding in the GGA. If PBE is used for both pseudo-
potential and valence states, the
a
and
b
lattice vectors are over - estimated and the
best estimate for the
c
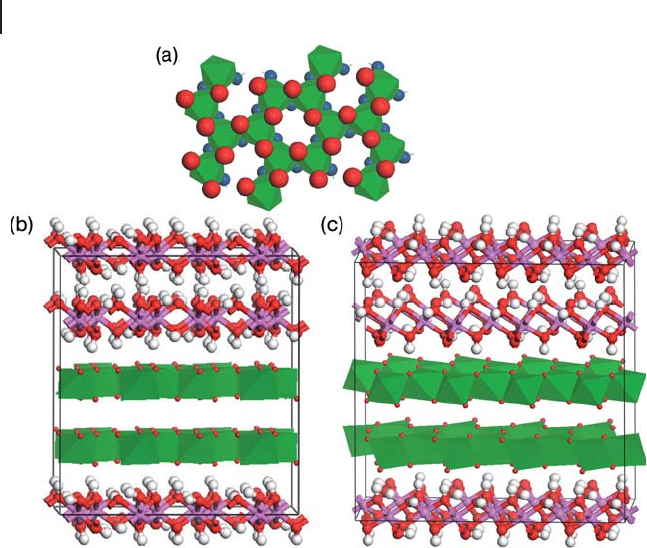
parameter is obtained. This structure is layered and held
together in the
c
direction by hydrogen bonding (see Figure 8.4 ). Using LDA/LDA
or LDA/PBE these hydrogen bonds are foreshortened, leading to underestimation
of the lattice vector. In the mixed case this means that a calculation using a GGA
in the valence states still performs like the LDA calculation because of the infl u-
ence of the pseudopotential. The use of mixed functionals has also been found to
lead to large errors in the calculated binding energies of molecules to metal sur-
faces which are corrected by employing a pseudopotential consistent with the GGA
used for the valence states [49] .
Table 8.3 Optimized structural parameters for Al(OH)
3
(gibbsite)
using various functionals for the pseudopotential/valence states.
parameter LDA/LDA LDA/PBE PBE/PBE Expt.
a)
a ( Å ) 8.504 8.623 8.798 8.684
b ( Å ) 4.867 5.017 5.110 5.078
c ( Å ) 9.224 9.598 9.674 9.736
β ( ° )
93.06 92.76 92.54 94.54
Cell vol. ( Å
3
) 381.2 414.7 434.4 428.0
Data taken from ref. [47] .
a Ref. [48] .
8.2 Electronic Structure Methods 341
342 8 Theory: Periodic Electronic Structure Calculations
8.2.3.4 Density of States
Within a band of states, the fact that
k
is virtually continuous means that the sepa-
ration between individual energy levels is vanishingly small. It makes no sense,
then, to try and draw a conventional energy level diagram of the type used in
molecular orbital calculations. The idea of a density of states ( DOS ) plot is to show
the number of states in a small energy interval as a function of state energy. The
number of states in a band per unit energy depends on the band width and the
number of cell - based functions that contribute to the band. The localized metal
cation d orbitals, for example, tend to give narrow bands with correspondingly
high density of states.
Figure 8.5 shows example calculated DOS plots for MgO and TiO
2
(rutile). In
both cases the zero of energy is taken as the highest occupied state which occurs
at the top of the upper valence band ( UVB ). The small tail on the UVB that indi-
cates some states with positive energy is an artefact of the smoothing process used
to construct the DOS plot from a calculation using fi nite number of
k
- points. In
addition to the total density of states, a decomposition into states associated with
Figure 8.4 The structure of Al(OH)
3
aluminum hydroxides: (a) a layer of edge -
sharing octahedra taken from the bayerite
structure, oxygen atoms on top of this bi - layer
shown as large red spheres while those on
the underside are shown as small blue
spheres. (b) The structure of bayerite: two
bi - layers are shown in the polyhedra
representation of (a) with H atoms omitted
for clarity, the remainder of the structure is
shown with Al in pink, O in red and H in
white. (c) The structure of gibbsite using the
same representations as (b).
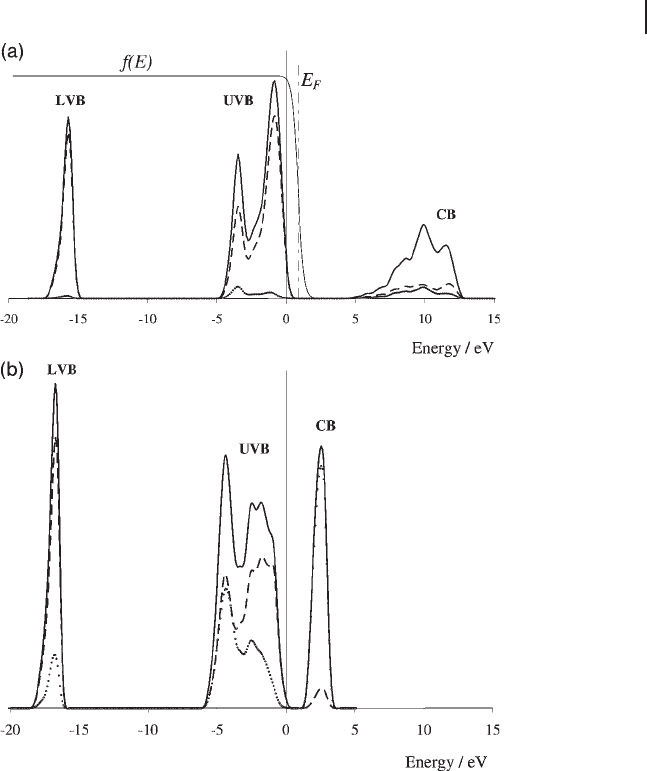
the oxygen and states associated with the metal atoms has been carried out. This
involves a projection of the density calculated using the plane - wave basis set onto
an atom localized basis of s, p and d functions. There is some arbitrariness in the
choice of this basis and so the absolute values of the partial density of states will
vary according to the choice made. In this case, we have relied on the PAW pseu-
dopotential functions used in the VASP code to defi ne the localized basis set into
Figure 8.5 (a) The DOS for MgO and the
smearing function ( f ( E )) used for calculating
the state occupancy. (This has a maximum
value of 1.) (b) The DOS of TiO
2
(rutile). Both
calculations used the VASP code with PAW
pseudopotentials and the PW91 functional; a
Gaussian smearing of 0.2 eV has been used
for each calculated point in the DOS. Total
DOS shown as a solid line, partial DOS for O
dashed line and for the metal ions dotted
line, arbitrary units are used for the DOS
magnitude. LVB, lower valence band; UVB,
upper valence band; CB, conduction band.
8.2 Electronic Structure Methods 343

344 8 Theory: Periodic Electronic Structure Calculations
which the projection takes place. These technicalities aside the relative contribu-
tion of metal and oxygen orbitals in each band do give some qualitative informa-
tion on the character of bonding in the materials.
The plot for MgO (Figure 8.5 a) is typical of a main group metal oxide which fi ts
with the classical ionic bonding model of oxide structures. The lower valence band
( LVB ) consists almost exclusively of O(2s) states and the UVB of O(2p) states. In
the conduction band ( CB ) both Mg and O basis functions contribute to the crystal
orbitals. The valence bands are completely fi lled and, since they have mainly O
character, this corresponds to complete transfer of valence electrons from Mg to
O to give the ionic species Mg
2+
and O
2 −
.
In the TiO
2
calculated DOS (Figure 8.5 b) the LVB is also mainly constructed
from of O(2s) orbitals but in the UVB the lowest energy states (between − 6 and
− 4 eV) are around 50 : 50 O and Ti in character with O(2p) orbitals being the major
constituent for higher energy states. In the conduction band the Ti orbitals make
up the larger part of the available states. The mixing of Ti and O states in the UVB
is consistent with a more covalent bonding model since electrons in these states
are shared between metal and oxygen atoms.
The width of the band formed is an indication of the degree of delocalization of
the band states and a comparison of Figure 8.5 a and b shows the biggest difference
in the UVB region. In the ionic model of MgO, the localization of electron density
on the O
2 −
ions leads to a relatively narrow UVB, while the covalent bonding in
TiO
2
gives rise to more delocalized states and so to a broader UVB.
This effect is also found for the bandwidth of the O(2p) bands for the alkaline
earth metal oxides which, at the LDA level of theory, decrease down the group
from a calculated value of 4.44 eV (MgO) to 1.83 eV (BaO). This is partly due to
the increase in lattice parameter, which spaces the O
2 −
ions more widely in BaO
than in MgO. However, in addition, it is found that the outermost valence elec-
trons for the metal ions interact more strongly with the O(2p) states in BaO than
in MgO, giving more localization of the electron density at O
2 −
and so a smaller
anion in BaO [50] .
Converging the self - consistent procedure in periodic calculations can be a diffi -
cult task, particularly if there are free states within any band. Movement of elec-
trons between almost identical states has little effect on the energy of the system
and so the search for the optimal distribution of electrons is hampered by many
trivial alterations to the occupation numbers. To make the process more effi cient,
partial occupancies can be used for states near the highest fi lled level by introduc-
ing a smoothing function which defi nes the occupancy as a function of state
energy, E
k
. The smoothing function used in the MgO calculation was:
fE
EE
k
k
()
=
−
()
+
1
1exp
F
σ
(8.24)
This expression is based on the statistical mechanics of electrons which have an
energy distribution given by Fermi – Dirac statistics [51] . The partial occupancy