Vlak J.M., de Gooijer C.D., Tramper J., Miltenburger H.G. (Eds.) Insect Cell Cultures: Fundamental and Applied Aspects
Подождите немного. Документ загружается.


242
allow direct processing of the whole broth. This elim-
inates the initial steps associated with cell separation
and speeds up the entire downstream processing.
Crude supernatant concentration
This step can be handled in very similar conditions
to the concentration of conditioned medium of mam-
malian cell origin (Menge et al., 1987; Tolbert & Prior,
1988; Goodall et al., 1992). For small scale operations,
concentration can be carried out by dead-end ultrafil-
tration in stirred cells with low protein capacity bind-
ing membranes (Goodall et al., 1992). Concentration
can also be achieved by ammonium sulfate precipi-
tation (Vissavajjhala & Ross, 1990). This technique
combines concentration and partial purification. An
alkaline pH shift to 8.0 also allows partial purifica-
tion by precipitating contaminating proteins and virus-
es (Hayman & Cox, 1994; Nutt et al., 1991). For
large scale, tangential flow ultrafiltration systems pro-
vide a large surface area of membrane. This tech-
nique has been applied successfully to the concentra-
tion of monoclonal antibody from hybridoma condi-
tioned medium (Prior et al., 1989). The membranes
are built in flat plate, spirals, hollow fibre or tubular
configurations. Some purification is also achieved by
size exclusion but in practice the resolution is poor.
Schlaeger et al. (1992) separated the cells by a con-
tinuous centrifuge and concentrated the 60 l culture
supernatant 10–20 fold by a tangential flow ultrafil-
tration system (Amicon SP20) with a filtration flow
rate around 100 l/hr. The molecular weight cut-off of
the hollow fiber membrane (2 10 kDa), allowed
complete retention of the secreted ectodomain of TNF
receptors (55 and 75 kDa). Concentration by ultra-
filtration can be combined with buffer exchange by
diafiltration which allows to reach the desired type of
buffer, ionic strength, and pH for the next step (Quelle
et al., 1989). It can be applied to affinity purification
in solution by using a high molecular weight affinity
ligand. The protein-ligand complex is thus retained by
an ultrafiltration membrane of high molecular weight
cut-off. Luong et al. (1987) described such a system to
purify trypsin from porcine pancreatic extracts. Affin-
ity purification can also be combined with ultrafiltra-
tion with membranes where the ligand is covalently
bound. Brandt et al. (1988) reported the purification of
Fibronectin from human plasma by gelatin covalent-
ly attached to cross-linked agarose and Nachman et
al. (1992) described membranes grafted with the sol-
uble domain of the IL–2 receptor for the purification
of recombinant IL–2 (mammalian cell origin). Kroner
et al. (1992) reported the purification of malate dehy-
drogenase from Escherichia coli and Saccharomyces
cerevisiae as a model system and Reif et al. (1994)
evaluated metal affinity membranes which should be
applicable to poly-His tagged proteins.
Cell breakage
Typically cell breakage techniques commonly used for
disruption of microorganisms have been applied to
the disruption of insect cells with little modification
(Table 1).
Homogenisation
One of the most common and simplest methods is
mechanical homogenisation. Due to the low shear
imparted, Dounce homogenisers have been the most
popular choice to homogenise insect cells in an ini-
tial step to recover recombinant protein (Lowery et al.,
1991; Chang et al., 1992; McGlynn et al., 1992; Chen
et al., 1993) Typically the cells are first subjected to
hypotonic shock (5–50 mM Tris-HCl, pH 7.6). The
hypotonic solution causes swelling of the cells which
renders them more susceptible to mechanical cell lysis.
The extent of cell breakage can be monitored by phase
contrast microscopy. Lysates obtained from homogeni-
sation protocols are usually clarified by centrifugation
at 10 000xg for 5 min.
Freeze-thaw
Freeze-thawing is a simple yet very efficient means of
cell lysis, however, the successful application of this
technique depends on the stability of the recombinant
protein after one or more consecutive freeze-thaws.
Wickham et al., (1991) successfully employed freeze
thawing with three different cell lines including Tn368,
Sf21 and Sf9 as a one step protocol for extraction of
Hink et al., (1991) applied the method for the
release of the same enzyme in 23 different cell lines.
Repetitive freeze-thawing has also been reported. Pre-
haud et al. (1990) disrupted Sf9 cells for purifying a
rabies virus nucleoprotein (N) by three cycles of freez-
ing in solid dry ice followed by immediate thawing at
37 °C. The cell debris was used as an immunogen for
the production of antibodies against the recombinant
protein in mice. Additionally freeze thawing has also
been used as a pre-treatment in combination with other
cell disruption steps (Chen et al., 1992; McGlynn et
al., 1992; Berndt & Cohen, 1990).
243
Osmotic shock
Hypotonic shock aids disruption of cells and can
cause osmotic damage to the nucleus and organelles
and is therefore very useful if the protein of inter-
est is located in these areas. Prehaud el al. (1990)
released intranuclear rabies nucleoprotein by resuspen-
sion of infected insect cell pellets in 0.5 mM Tris-HCl
(pH 7.5) on ice for 30 minutes. Similarly Giner et
al. (1992) achieved one step lysis using a hypotonic
buffer for the release of full-length human poly(ADP-
ribose)polymerase (PARP) in baculovirus infected Sf9
cells.
Sonication
Sonication (ultrasonic disintegration) has been used
mainly as one step in a cascade of other insect cell
disruption steps to ensure cell lysis (Graber et al., 1992;
Wang et al., 1994; Denis et al., 1991). The cells are
sonicated continuously or with a number of pulses of
sonic energy of a few Watts (Stauffer et al., 1991; Peng
et al., 1993) after other homogenisation or freeze thaw
procedures. Baylis et al. (1991) used hypotonic buffer
followed by gentle sonication at 0 °C using a water
bath sonicator to release EBV alkaline Dnase from
Sf9 cells. A combination of flash freezing at –80 °C,
probe sonication in hypotonic buffer and centrifugation
followed by a second round of sonication was required
to release protein kinase-C from insect cells (Rankl et
al
.,
1994). Using sonication as a one step process would
require careful consideration of parameters including
the duration and number of consecutive pulses as well
as the distribution of the integrity of membrane states
found at various times post-infection within the cell
population.
Gas cavitation
Gas cavitation has also been reported for disruption of
baculovirus infected insect cells. Typically this proce-
dure involves the suspension of cells in a homogeni-
sation buffer in a nitrogen containing metal cylinder
for 5–30 min at high pressures (600 psi and above).
On return to atmospheric pressure, the nitrogen gas
dissolved in the cytoplasm is released aiding cell dis-
ruption by rapid expulsion of the cells from the cylin-
der via a narrow orifice. Typically organelles, includ-
ing the nucleus, are released intact using this method.
Graber et al. (1992) thawed frozen harvested cells into
homogenisation buffer and burst them by cavita-
tion (300–600 psi for 20 min). The intact nuclei and
cell debris were removed by low speed centrifugation
leaving the soluble cell extract that contained two sub-
units of recombinant G proteins. Tang et al. (1991) also
244
employed the procedure of cavitation but retained
the cell membrane pellet for mechanical homogeni-
sation, followed by detergent extraction, to release a
recombinant calmodulin-activated (Type 1) adenylcy-
clase.
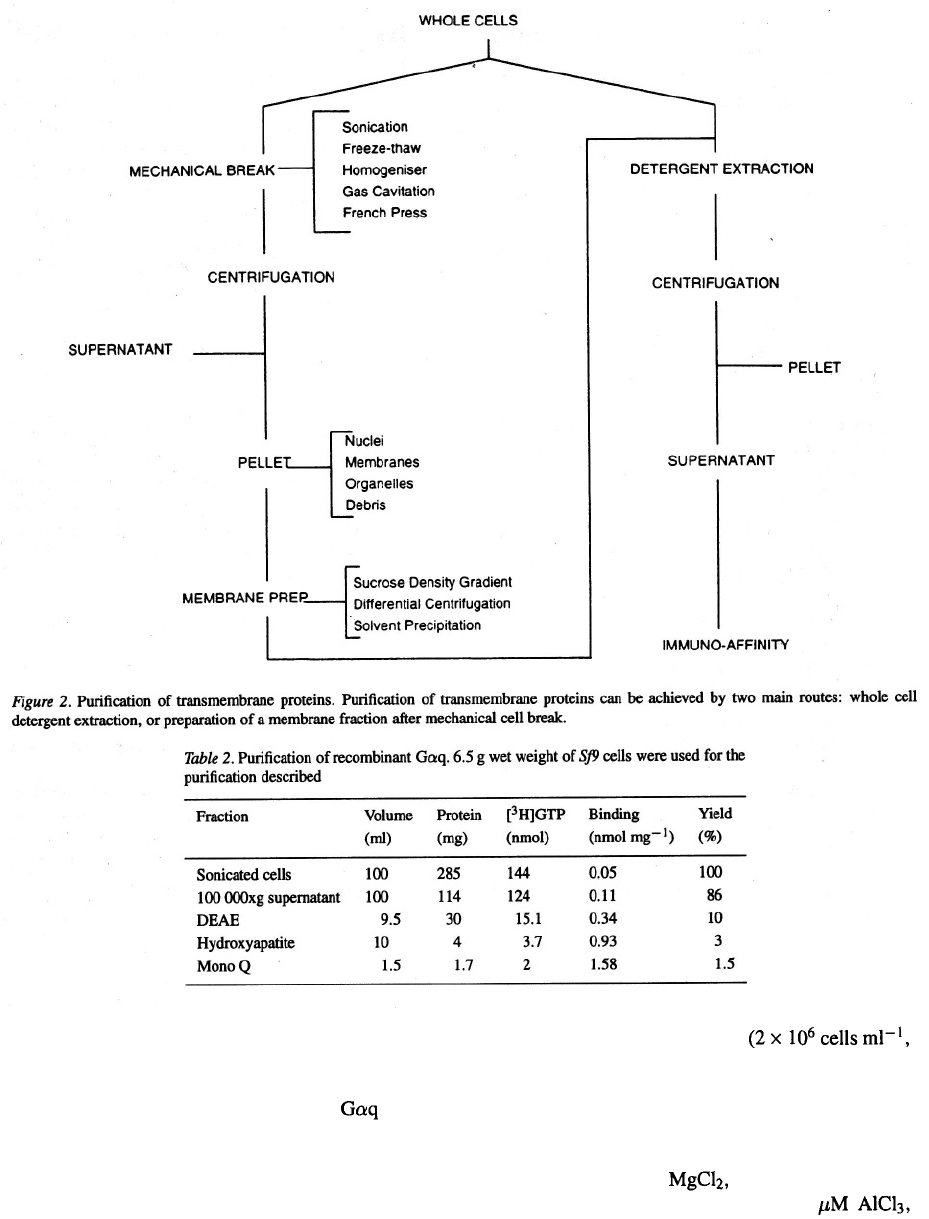
Cell breakage for membrane proteins
Disruption of insect cells for the purification of mem-
brane proteins involves special techniques. There are
basically two ways to purify transmembrane receptors
from recombinant insect cells (Figure 2).
1) Preparation of membranes from cell lysates fol-
lowed by detergent extraction.
Separation of the membrane fraction is achieved by
continuous or discontinuous sucrose density gradi-
ent (Stauffer et al., 1991) or by ethanol precip-
itation (Pochon et al., 1992). Alternatively, cell
lysis is followed by differential centrifugation of
the membrane components (Evans, 1978; Sheeler,
1981; Parker et al., 1991; Reilander et al., 1991).
For these methods, based on the enrichment of the
preparation with membranes, the aim is to conserve
the membranes integrity as much as possible in a
homogeneous preparation. In all cases the prepara-
tion starts by cell breakage followed by a centrifu-
gation step which isolates the insoluble fraction
containing the membranes.
2) Whole cell detergent extraction.
This method combines breakage and membrane
protein extraction in a single step. Cationic, anion-
ic or zwitterionic surfactants or detergents have
been used for recovery of intracellular proteins of
the refractile bodies and cytoplasmic aggregates
found in recombinant bacterial cultures. The same
has been applied to insect cell cultures. Researchers
have employed the process of surfactant solubilisa-
tion as the primary step in purification and isolation
of some membrane associated proteins (Domingo
& Trowbridge, 1988; Peng et al., 1993). Proteins
have been extracted from the membrane lipid bilay-
er of baculovirus infected insect cells by inser-
tion of their hydrophobic regions into detergent
micelles. Wells & Compans (1990) purified the
envelope glycoprotein of HIV (gp160) and used a
lysis buffer including l% triton X–100, l% Deoxy-
cholate and 0.1% SDS as detergents. Pelleted cells
were resuspended in this buffer, incubated on ice
for 15 min and microfuged before being used for
immune precipitation. Similarly, Landolfi et al.
(1993) used surfactant solubilisation for the initial
purification of herpes simplex virus type 2 glyco-
protein D.
Some published examples describe cell disruption
by detergents only (Narum et al., 1993; Webb et al.,
1989; Paul et al., 1990). The detergent cell extract
contains both the insoluble and the soluble proteins
and subsequently requires additional purification
steps to achieve good purity of the recombinant
membrane protein. A disadvantage associated with
detergents is that membrane-bound proteases may
be activated during prolonged procedures, although
the effect has not yet been reported in insect cells.
Part 2: Protein specific processes: case studies
This part illustrates purifications of proteins routine-
ly carried out at large scale in our laboratories. They
were chosen to represent three different types of pro-
teins namely cytoplasmic, secreted and embedded in
the cytoplasmic membrane. The aim is to detail the
experimental procedures used and the results obtained
in each case. These methods should be equally applica-
ble to other recombinant proteins.
Case study 1: model of cytoplasmic protein
purification
Heterotrimeric guanine nucleotide-dependent regula-
tory proteins (G proteins) are an essential part of
the signal transduction pathways which mediate the
cell’s response to hormones and neuromodulators. The
Gq class of G proteins includes from mouse,
drosophila and squid photoreceptor membrane. Mem-
bers of this class mediate the hormonal stimulation
of membrane phosphoinositides by
and (Smrcka et al., 1991; Gutowski et al.,
1991; Taylor et al., 1991). A variety of G protein
subunits have been purified from baculovirus infect-
ed insect cells by several groups (Graber
et al.
, 1992;
Robishaw et al., 1992; Labrecque et al., 1992; Hep-
ler et al., 1993; Jones et al., 1993; Ueda et al., 1994;
Singer et al., 1994). We had previously purified
from mouse brain and shown it to stimulate the activ-
ity of polyphosphoinositide-specific Phospholipase C
The purpose of this work was to obtain
large amounts of protein from recombinant sources for
futher studies and characterization. cDNA from
mouse brain (Strathmann & Simon, 1990) was cloned
into a baculovirus expression vector downstream of the
polyhedrin promotor using standard methods (Chapter
245
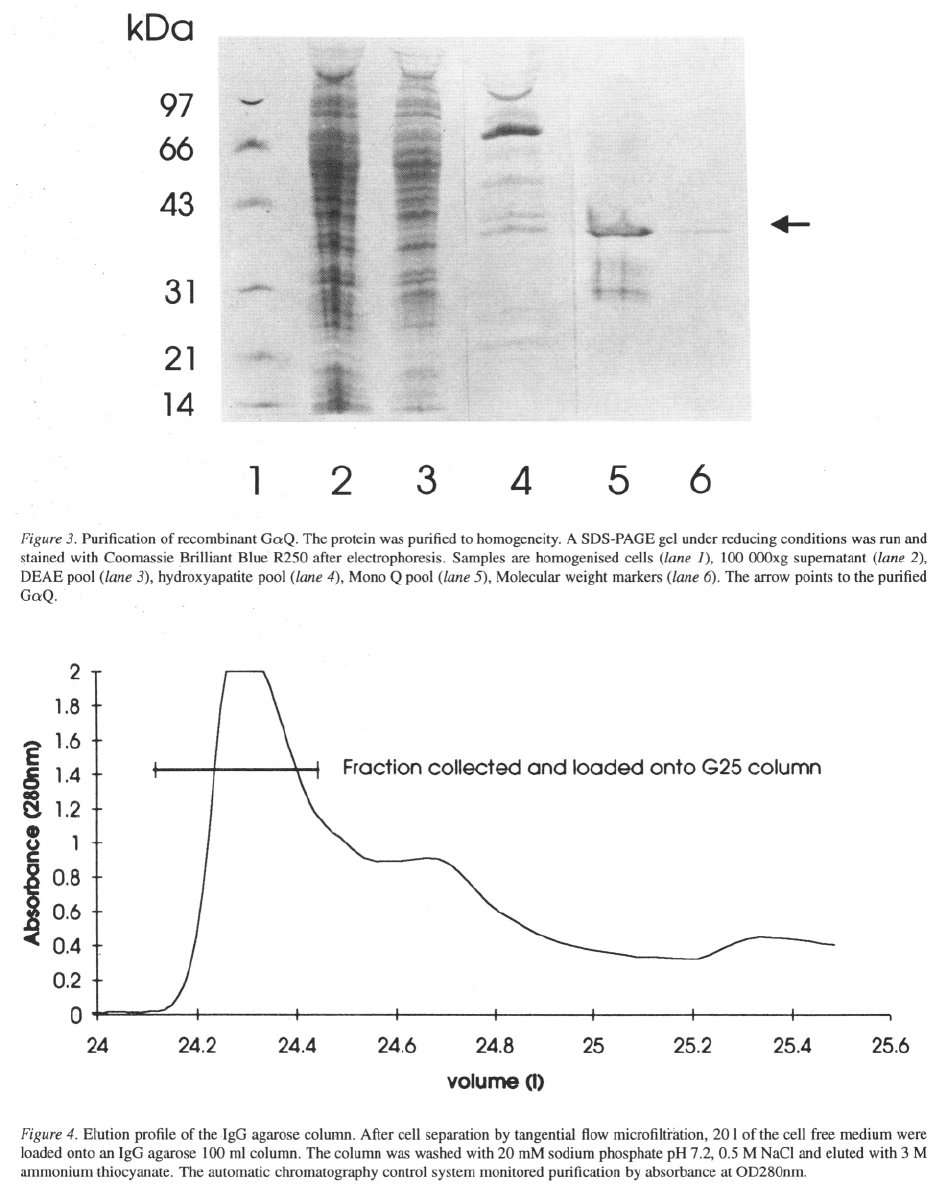
9, this issue). Upon infection of Sf9 cells, a 42 kDa band
showed up on a SDS-PAGE gel stained with Coomassie
blue. Western blot analysis of uninfected and infected
cells showed the presence of endogenous in Sf9
cells which has made the purification and characteri-
sation of the recombinant protein more difficult.
The cell extraction protocol is modified from that of
Graber et al. (992). All the extraction steps were done
at 4 °C. Cells in the culture broth
4 or 15 litre reactors) were harvested by low speed
centrifugation (200xg, 15 min). The resulting pellet
was immediately frozen at –80 °C and stored for a
few days. The cells were thawed in 15 × their wet
weight of homogenisation buffer (10 mM Tris-HCl,
25 mM NaCl, 10 mM 1 mM EDTA, 1 mM
DTT, 0.1 mM PMSF, 10 mM NaF, 10
246
10 GDP, pH 8.0) and broken by 2
×
30 s sonica-
tion. GDP was included in the homogenisation buffer
to keep the protein correctly folded and stable. Soni-
cated cells were centrifuged for 90 min at 100 000xg
to remove unbroken nuclei and cell debris. The solu-
ble cell protein fraction was loaded onto a 2.5 × 5 cm
DEAE column equilibrated in TED buffer (50 mM
Tris-HCl, 0.02 mM EDTA, 1 mM DTT, 1 mM
pH 8.0) and eluted with a gradient from 0–0.7 M NaCl
in TED using a flow rate of 1.2 ml Column
fractions were assayed for GTP binding activity using
Each assay tube contained 10 sample in a
total volume of 150 incubation buffer (75 mM Na-
HEPES, pH 8.0, 50 mM 100 mM NaCl, 1 mM
EDTA, 0.5 mM ATP and 1 GTP (4 of
GTP ). Tubes were incubated for 20 min at 30 °C
and binding terminated by addition of 1 ml ice-cold
wash buffer (50 mM Tris-HCl, 100 mM NaCl, 1 mM
EDTA, 25 mM pH 8.0) followed by rapid filtra-
tion through nitrocellulose filters. Those fractions with
binding activity were pooled, diluted with 4 volumes of
buffer containing 10 mM 10 mM Tris-HCl,
pH 8.0 and loaded onto a 1.5×4 cm hydroxyapatite
column equilibrated in the same buffer. The column
was eluted from 0–300 mM with a 80 min
linear gradient and a flow rate of 0.8 ml The
fractions containing GTP binding activity were load-
ed onto a HR 5/5 MonoQ column equilibrated with
25 mM Tris-HCl, 0.02 mM EDTA, 1 mM DTT and
1 mM MgCl
2
and eluted from 0–1 M NaCl for 100 min
with a flow rate of 0.8 ml The proteins were
well separated and the GTP activity could be traced to
one fraction. The protein was purified to homogeneity
as shown on a SDS-PAGE gel stained with Coomassie
Blue (Figure 3). However, because of the number of
steps involved in our purification scheme, the final
yield was low and on average 10 fold less than pub-
lished data (Graber et al., 1992). The activity of the
pure Gaq protein was confirmed by a PLC activation
assay using the method described by Gutowski et al.
(1991).
Case study 2: E-selectin, model of a secreted protein
E-selectin is a cell adhesion molecule which mediates
the initial ‘rolling’ of neutrophils on endothelium at a
site of inflammation by interaction with the tetrasac-
charide ligand sialyl Lewis X presented on endothelial
cells (Springer & Laski, 1991). We previously report-
ed (Cavegn et al., 1992) the production of a recom-
binant form of the protein in which the C-terminal
membrane-spanning region was replaced with two con-
sensus domains from Staphylococcus aureus protein A
termed ‘zz’ (Löwenadler et al., 1987).
The modified E-selectin (E-selectin-zz,) was
cloned into baculovirus to give a secreted protein, and
after culture was purified using immobilised IgG to
capture the protein by interaction betweeen the Fc
domain of IgG and the zz domains of E-selectin-zz.
Trichoplusia ni cells (which were adapted to grow
in suspension culture) were cultured in a 36 litre air-
lift fermenter using Excell 401 medium supplemented
with 5% foetal calf serum. The cells were infected
at a density of cells with baculovirus
(Multiplicity of infection = 1), and harvested 3 days
after infection ( cells 93% viable).
The cells and the medium (36 litres) were separat-
ed using a tangential flow membrane apparatus (Mil-
lipore Prostak Dual-pump mammalian harvest sys-
tem) fitted with three 10-stack modules (PSGVAG101,
0.22 ) of 0.84 each. The recirculation rate was
6.7 1 the permeate flow was 0.63 1 (flux
15 1
and the transmembrane pressure was
0.2 Bar. After washing the retentate with phosphate
buffered saline, the total volume of clarified super-
natant was 40 litres. A cocktail of protease inhibitors
(benzamidine 1 mM, PMSF 1 mM, leupeptin 5
SBTI 5 and pepstatin A 5 ) and sodium
azide (5 ) were then added to prevent proteolysis
and bacterial growth, and the supernatant was stored at
4 °C prior to processing.
The E-selectin-zz was purified (room temperature
throughout) in two 20 litre batches using a Quantasep
instrument (Sepragen). The supernatant was loaded
onto human IgG-agarose (from ACL, packed in a
100 ml radial flow column) at a flow rate of 100 ml
After the column had been loaded and washed
with buffer (20 mM sodium phosphate pH 7.2, 0.5 M
NaCl) the E-selectin-zz was eluted with 3 M ammoni-
um thiocyanate. The pooled material was then desalted
on a 3 1 Sephadex G25 column using sodium phos-
phate buffer (without NaCl) at a flow rate of 300 ml
The entire purification was run unattended by
using the chromatography control software. The purity
of the protein was determined by gel electrophoresis.
Adhesion of HL60 cells to E-selectin-zz bound to IgG
coated wells was used to monitor biological activity.
The elution profile from the IgG column is shown
in Figure 4. The pooled material was loaded directly
onto the G25 column to remove the ammonium thio-
cyanate. This compound absorbs strongly at 280 nm
and is responsible for the absorbance after the pro-
247
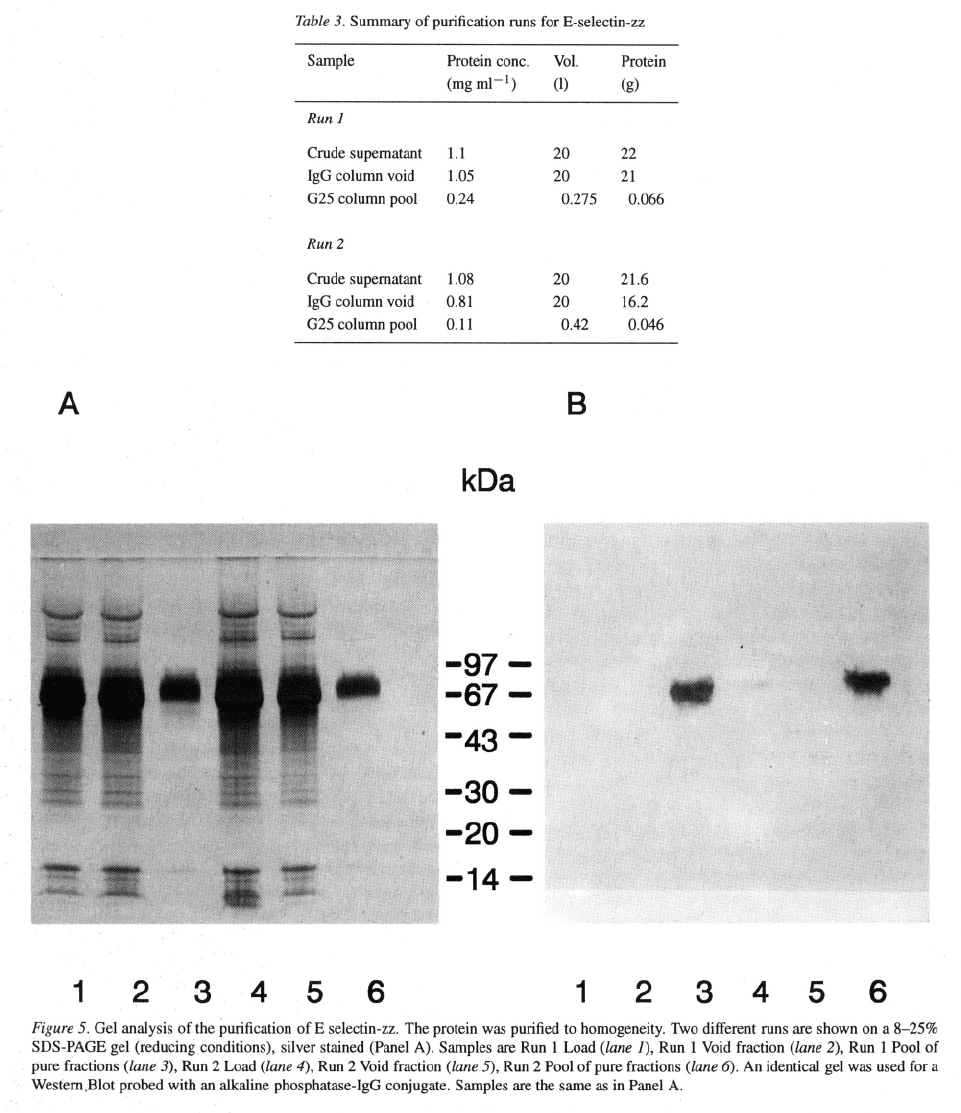
248
tein peak has eluted. The purification is summarised
in Table 3 and the analysis by gel electrophoresis and
silver staining is shown in Figure 5 (panel A). The
main protein band in the load and void fractions was
albumin derived from the culture medium. Analysis of
the column fractions on an 8–25% reducing SDS gel
(Pharmacia Phastgel) followed by silver staining indi-
cated that the protein in the final pool was >95% pure.
249
On a similar gel, the proteins were transferred to PVDF
membrane and probed with an alkaline phosphatase-
IgG conjugate (Figure 5, panel B). E-selectin-zz could
be detected as a weak band in the load sample, but not
in the void fraction. As expected, a strongly staining
band could be seen in the pool. Using the biological
assay, the E-selectin-zz activity could not be detected
in the load (or the void) fraction, but in the G25 pool
activity diluted out to 1/128 without loss of signal.
The identity of the protein from a similar purifica-
tion scheme was confirmed by N-terminal sequence
analysis and this also showed that the signal pep-
tide was fully removed. The mass of the protein as
determined by MALDI-TOF spectroscopy was cen-
tred around 70 kDa, but the signal peak was broad,
indicating sample heterogeneity which was later con-
firmed to be due to glycosylation variants (data not
shown). The two step purification scheme, involving
IgG capture then desalting was shown to give materi-
al suitable for high throughput screening, biochemical
studies and structural work (Cooke et al., 1994).
This study shows the power of using an affinity
tag for the purification of a secreted protein. Signifi-
cant quantities of the E-selectin-zz were purified using
an extremely straightforward purification scheme. The
scheme was automated, and the scale was matched
conveniently to the fermentation. The E-selectin-zz
obtained was active in the biological assay, was >95%
pure and usable for all the applications required. The
same purification strategy was applied to the recovery
of other adhesion molecules (ICAM, VCAM) fused to
zz domains.
Case Study 3: Purification of CD23, model of a
transmembrane protein
CD23 is a type II single transmembrane receptor found
at the surface of many hemopoietic cells. It is the low
affinity receptor for IgE but other biological activities
related to B cell proliferation and antigen presentation
have been described for this molecule (Aubry et al.,
1992). In our search for its ligand, now identified as
CD21, we had to purify CD23 from infected Sf9 cells
on a large scale for incorporation into fluorescent lipo-
somes. In order to identify CD21 as the CD23 ligand,
many cell lines were screened for their ability to bind
to CD23 liposomes (Pochon et al
.,
1992).
Most of the published work on transmembrane pro-
teins expression in insect cells focuses on the expres-
sion and the characterisation of recombinant recep-
tors on insect cell surface. Membrane preparations or
whole cell extracts have been used without any purifi-
cation for functional or binding studies. This kind of
approach is only suitable for high affinity receptors
and high density expression at the cell surface (Jensen
et al., 1992; Greenfield et al., 1988). Characterisa-
tion of in vitro activity of detergent solubilized trans-
membrane proteins is also possible when the protein
contains a domain that possess enzymatic activity (Li
et al
.,
1992). Other activity characterisation studies
require a highly purified receptor and reconstitution of
the activity in artificial membranes (Parker et al., 1991;
Reiländer et al., 1991). When the recombinant recep-
tor is expressed at very low level at the cell surface, any
contamination by other proteins will have a dispropor-
tional effect on the purity. To obtain the best purity, it is
advisable to start from an enriched membrane fraction.
Therefore our strategy was to first disrupt the insect
cells and prepare a fraction enriched in plasma mem-
branes. To reduce the number of purification of steps
we used affinity chromatography in detergent with an
anti-CD23 antibody. The protein was then reconstitut-
ed in artificial membranes by incorporation into lipo-
somes.
Cell disruption
Insect cells were found more difficult to break than
mammalian cells for our purpose. Breakage by soni-
cation or by mechanical shearing, routinely used for
mammalian cells, was not sufficient to break all the
cells. We obtained a very good breakage efficiency
using a French pressure cell under the same conditions
used to break Escherichia coli cells (Wingfield et al.,
1987). Fresh or frozen cells were suspended in absence
of detergent in hypotonic buffer, 10 mM Tris/HCl pH
7.8, with protease inhibitors (1 mM TLCK, 1 mM ben-
zamidine, 1 mM PMSF, 10 mM iodoacetamide). The
cell suspension was then passed two or three times
through a French pressure cell at 18 000 psi. Cell
breakage was monitored by microscopic observation
after addition of 0.1% Trypan blue. The cell lysate was
then diluted two fold in 10 mM Tris/HCl pH 7.8 buffer
containing 0.1M NaCl, protease inhibitors (same com-
position as above) and 0.5 M sucrose and the solution
was centrifuged at 20 000xg for 60 min and supernatant
was discarded.
Preparation of an enriched membrane fraction
Four methods (sucrose density gradient, differential
centrifugation, Triton X-114 phase partitioning and
ethanol precipitation) were tested to purify the recom-

250
binant transmembrane CD23 receptor from Sf9 insect
cells. They all aimed at a preparation enriched in mem-
branes, starting form the cell lysate pellet. After each
preparation, the membrane fraction was purified by
immune-affinity as described by Pochon et al. (1992).
Briefly, the membranes were extracted in 1% Triton
X–100 detergent and the insoluble material was sepa-
rated by centrifugation at 150 000xg for 60 min. The
supernatant was immunopurified on a MAb25-Affigel
10 column (MAb25, an anti-CD23 monoclonal anti-
body, was coupled at 3 mg per ml resin) with con-
stant recycling overnight. The immunoaffinity column
was washed with PBS containing 1% Triton X–100
and 100 mM NaCl, then with PBS buffer contain-
ing 0.1% Triton X–100. In the third wash, the Triton
X–100 detergent was replaced by 50 mM
glucopyranoside (OGP), a dialysable detergent. CD23
was finally eluted with PBS buffer pH 6.5 contain-
ing 50 mM OGP,and 3 M The protein was
desalted and further purified on a Superdex–200 gel
filtration column equilibrated in PBS buffer containing
50 mM
OGP.
Sucrose density gradient
Centrifugation of the cell lysate on a sucrose density
gradient yielded two fractions which were enriched in
CD23 membranes: phase 1 in 28% w/w sucrose and
phase 2 in 42% w/w sucrose. Both phases were diluted
separately, centrifuged and extracted with 1% Triton
X–100, prior to affinity purification. Analysis by SDS-
PAGE and western blotting using a polyclonal anti-
CD23 antibody showed that the protein isolated from
phase 1 had a greater degree of purity than that isolated
from phase 2 (data not shown). The receptor was then
incorporated into fluorescent liposomes and the activity
measured by the ability of the liposomes to bind to the
RPMI 8226 cell line expressing CD21 at its surface.
Both preparations were active. Using this method, the
purification yield was around 18 cell culture
(10
9
cells ). The major problem with this method
was the poly-dispersion of the membrane vesicles over
the gradient. Although we obtained a slight enrichment
of the phases 1 and 2 with CD23 positive membranes,
the rest of the gradient also contained traces of CD23
receptor which were discarded.
Membrane preparation by differential centrifugation
This method is routinely used with mammalian cells
to isolate membranes from specific cell compartments
and was reviewed by Findlay & Evans (1987). Several
membrane enriched fractions at various densities were
obtained, and individually extracted with 1% Triton
X–100. The insoluble material was removed by cen-
trifugation and the soluble extract was affinity puri-
fied as described above. The western blotting analysis
of the various fractions shows that CD23 was found
distributed in all cell compartments, both in unbro-
ken cells and in isolated nuclei, endoplasmic reticulum
and plasma membranes. Using this method, the final
purification yield was 7 cell culture ( cells
).
Phase partitioning in Triton X–114
Triton X–114 detergent aggregates into micelles when
the temperature is raised above 20°C. This property
was exploited to isolate a membrane rich fraction from
the cell lysate. Insect cells were lysed at 4 °C with
a buffer containing 1% Triton X–114 and the insol-
uble material removed by centrifugation (100 000xg
30 min). Upon heating the extract at 37 °C for 10 min,
two phases formed. After centrifugation, the cloudy
detergent phase enriched in membrane proteins was
collected (Bordier, 1981). This method did not result
in any purification of CD23 as judged by SDS-PAGE
and Western Blot analysis. It was thus not pursued
further.
Whole membrane preparation by ethanol
precipitation
The cell lysate pellet was washed by several centrifu-
gations (20 000xg, 60 min) in 10 mM Tris/HCl pH
7.8 buffer containing 0.1 M NaCl, protease inhibitors
(and 0.5 M sucrose. The pellet was resuspended in
PBS containing 1 mM 1 mM and pro-
tease inhibitors (same composition as above). It was
then precipitated with an equal volume of cold ethanol
(–20 °C), producing a clean membrane fraction. The
process was repeated twice. The membrane pellet was
extracted in Triton X–100 as described below and the
CD23 further purified by immuno-affinity chromatog-
raphy. Despite the denaturing potential of ethanol, no
loss of activity in these preparations could be detected
when working at 4 °C. On average, the purity of the
final product was around 95% as measured by gel elec-
trophoresis (Figure 6). The yield was around 120
CD23 culture which was at least 6 fold higher than
that obtained by the sucrose density gradient technique.
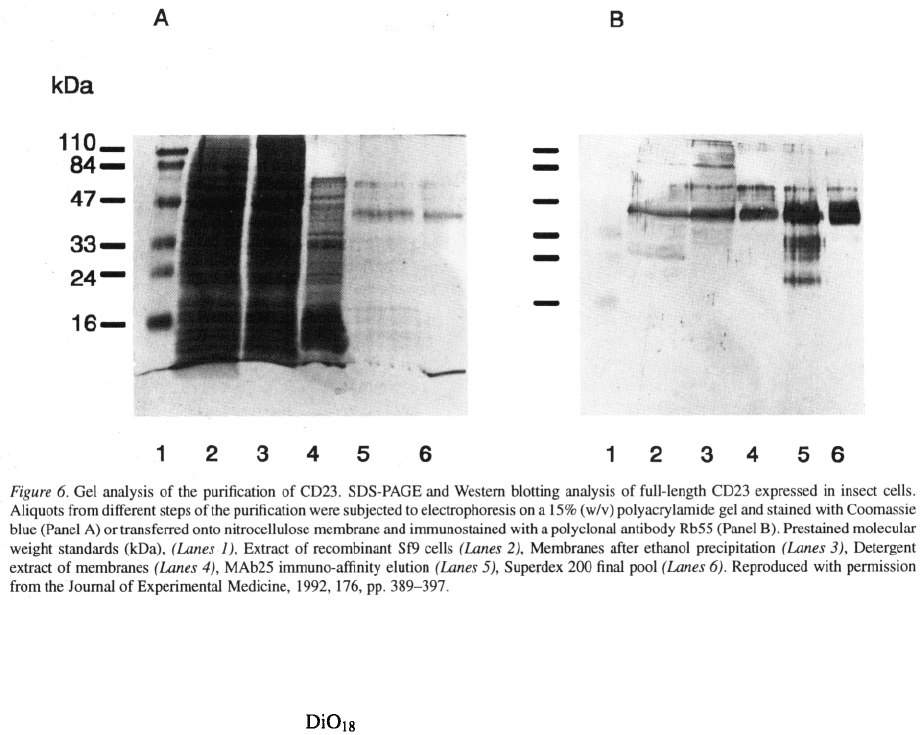
251
Preparation of liposomes and activity testing
The purified CD23, solubilized in 50 mM OGP, was
further incorporated into fluorescent liposomes made
of synthetic POPC and fluorescent phospho-
lipids solubilized in OGP (Pochon et al., 1992). The
detergent was then dialysed and single layer lipo-
somes were formed which contained the transmem-
brane CD23. Control liposomes were made with gly-
cophorin A protein by the same method. Recombinant
CD23 incorporated into fluorescent liposomes was able
to bind its ligand, CD21, on selected T and B cell lines
(Figure 7). Competition of the binding with a panel of
antibodies (Aubry et al
.,
1992) proved the specificity
of the interaction.
A summary of the results for each method is pre-
sented in Table 4. In conclusion, this study demon-
strates that the baculovirus/insect cell protein expres-
sion system is suitable to production of native, func-
tional transmembrane proteins. Purification of trans-
membrane proteins from insect cells can be achieved
using essentially the same methodology as for mam-
malian cells. Affinity purification in detergent is a
very powerful method for receptor purification. The
best results were obtained starting from a membrane
enriched fraction prepared by ethanol precipitation.
Part 3: Key issues for future developments
Downstream processing for the recovery of prod-
ucts from the baculovirus/insect cell system has been
reported to be essentially similar to that involved in
mammalian systems. There appear to be however sev-
eral unique problems, situations and myths which arise
when purifying products from this multicomponent
system.
The key issue involved in cell separation and dis-
ruption as preliminary isolation steps is maintenance
of compartmentalisation of the product. The litera-
ture has indicated the diversity of product localisation
and therefore the need to maintain discrete boundaries
during product isolation. Regimes involving complete
membrane and organelle disruption are far from opti-
mal and new protocols respecting compartmentalisa-
tion need to be developed.
Apart from cell protease release, shear sensitiv-
ity and alteration of the product during purification