Ramanathan Sh. (Ed.) Thin Film Metal-Oxides: Fundamentals and Applications in Electronics and Energy
Подождите немного. Документ загружается.

9 Applications of Thin Film Oxides in Catalysis 283
9.2 Tuning Electronic Properties of Novel Metal Oxide
Nanocrystals Using Interface Interactions: MoO
3
Monolayers on Au(111)
9.2.1 Background and Introduction
MoO
3
is an important catalyst in the reduction of nitrous oxides [21] and in the
partial oxidation of alkanes [22]. The thermodynamicallystable form of bulk MoO
3
,
known as ’-MoO
3
, consists of bilayers parallel to the (010) plane, bonded through
weak electrostatic interactions. Each bilayer consists of two monolayers of distorted
MoO
6
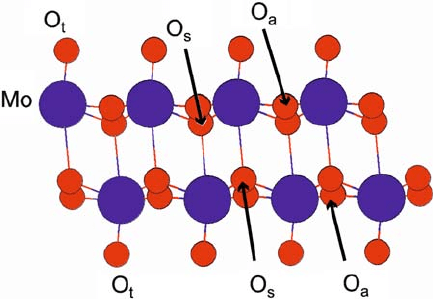
octahedra, with three distinct oxygen species (Fig. 9.1). The asymmetric
bridging oxygen O
a
is collinear between two Mo atoms, forming one long and one
short bond with each of them. The symmetric bridging oxygen O
s
forms two bonds
of equal length to two Mo atoms of one monolayer and one elongated bond to an-
other Mo atom in the other monolayer of the bilayer. Finally, each terminal oxygen
O
t
is bonded to one Mo atom. The catalytic activity of MoO
3
depends sensitively
on the type of oxygen species exposed [23].
To elucidate the relative importance of these distinct oxygen species in cataly-
sis, experiments have been performed on thin molybdenum oxide films obtained by
oxidizing Mo(110) [1,21,24]. By varying the conditions of oxidation, the structure
of the surface oxide can be controlled. These studies have shown that the catalytic
activity of molybdenum oxide is intimately related to its structure, including the
concentration of oxygen vacancy defects [1, 21, 24]. Using DFT calculations on
bulk MoO
3
, we have further elucidated the importance of distinct oxygen species
and of vacancies in the catalytic process [3, 25, 26]. For example, we have found
Fig. 9.1 (color online) Atomic structure of bilayer in bulk MoO
3
. The Mo atoms are shown as
larger spheres and the O atoms as smaller spheres; the three inequivalent positions of O atoms are
indicated and labeled O
s
(symmetric bridging), O
a
(asymmetric bridging), and O
t
(terminal).
284 S.Y. Quek and E. Kaxiras
that terminal oxygen vacancies in bulk MoO
3
results in an exothermic C–H bond
breaking reaction in methane, with H preferably binding to terminal oxygen sites
and CH
3
to vacancy sites [3].
Here, we show that MoO
3
nanocrystals grown on Au(111) have distinct elec-
tronic and structural properties from bulk MoO
3
, and we suggest the possibility of
tuning the catalytic properties of MoO
3
and other ultrathin oxide films using inter-
face interactions [27]. In contrast to the bilayered structure of the bulk oxide crystal,
MoO
3
nanocrystals grown on Au(111) are only one monolayer thick with the Au
surface acting as the other half of the bilayer by satisfying local bonding require-
ments through charge redistribution at the interface. Epitaxy with the Au lattice is
achieved through the ability of the Mo–O bonds to rotate about one another, because
dihedral angles represent a relatively soft degree of freedom in transition metal ox-
ide lattices. The oxide layer becomes semimetallic as it strains to enhance bonding
with the Au substrate. These distinct electronic properties result in chemical proper-
ties that are different from bulk MoO
3
, as illustrated by the energy of H adsorption
at various surface sites.
MoO
3
nanocrystals were grown on Au(111) surfaces by both chemical (CVD)
and physical vapor deposition (PVD) of Mo, followed by oxidation using NO
2
.
In the CVD experiments, the surface was typically exposed to 1 L of Mo.CO/
6
and 10 L of NO
2
alternatively at 450 K, followed by annealing to 600 K for 1 min
after every 4 cycles of dosing, for a total of 16 cycles. The PVD syntheses were
performed at 450–600 K. Typically, 0.3 monolayers (ML) of Mo was deposited at
a flux of 0:25–0:75 ML=min and oxidized by exposure to 20L of NO
2
.Further
experimental details are described elsewhere [28].
High-resolution scanning tunneling microscopy (STM) and low-energy electron
diffraction studies indicate that the MoO
3
islands grown by either technique have a
c.4 2/ unit cell. While bulk MoO
3
consists of weakly interacting bilayers [29],
STM images reveal that the islands are one monolayer in height, which corresponds
to half of the height of the bilayer found in bulk MoO
3
(Fig. 9.2b). This interesting
surface structure has another important ramification: although clean Au(111) has
a herringbone reconstruction [30], STM images indicate that the reconstruction is
lifted under the molybdenum oxide islands (Fig. 9.2a), a feature we adopt in the
theoretical model of the system.
9.2.2 Computational Details
The atomic and electronic structure of this system were studied using DFT, with the
projected augmented wave method [31] and the Perdew-Wang 91 generalized gra-
dient approximation for the exchange-correlation functional [32], as implemented
in VASP (Vienna Ab-Initio Simulation Package) [33]. We use a slab model with six
Au layers in a c.4 2/ supercell, separated by 16.5
˚
A of vacuum before the oxide is
introduced. The MoO
3
monolayer and the top three Au layers were relaxed until the
magnitude of forces on all atoms was less than 0.01 eV/
˚
A. Geometry optimizations
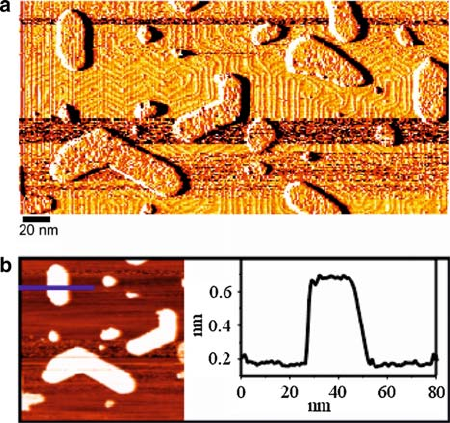
9 Applications of Thin Film Oxides in Catalysis 285
Fig. 9.2 (color online) STM images of MoO
3
islands on Au(111). The sample was prepared by
PVD of 0.3 ML of Mo on Au(111) at 600 K, and subsequent oxidation by exposure to 20 L of
NO
2
at 600 K. The images were collected at room temperature at a sample bias of C2:0 Vanda
tunneling current of 0.15 nA. (a) Constant height image. The Au herringbone reconstruction runs
parallel to the straight island edges, and bends sharply at rough island edges, which indicates that
the Au(111) reconstruction is lifted under the MoO
3
islands. (b) Corresponding constant current
image of a portion of (a). The line scan on the right is taken along the horizontal bold line in the left
panel and shows that the MoO
3
island has an apparent height of 0.5 nm, in contrast to the height
of a bulk bilayer cell which is 1.39 nm [29], thus suggesting that the island consists of a MoO
3
monolayer.
were performed using a plane-wave cutoff of 400 eV and a 3 3k-point mesh. As
a convergence check, a 6 6k-point mesh did not change the optimized geometry
significantly. Total-energy differences and electronic charge densities were calcu-
lated using a plane-wave cutoff of 500 eV and a 12 12 k-point mesh. Such a mesh
gave converged total energies in a bulk-terminated Au(111) surface.
9.2.3 Results and Discussion
Our calculations reveal that the MoO
3
monolayer (Fig. 9.3a) distorts to fit the Au lat-
tice and has distinct symmetry properties from its bulk analogue (Fig. 9.3d), which
served as the guide for the initial oxide structure. In contrast to the bulk case, the
MoO
3
monolayer has two nonequivalent planes of reflection and glide symmetry.
The slab appears to be composed of MoO
3
units tilting alternately forward and back-
ward relative to the surface normal, along the axes of reflection (Fig. 9.3b). Using
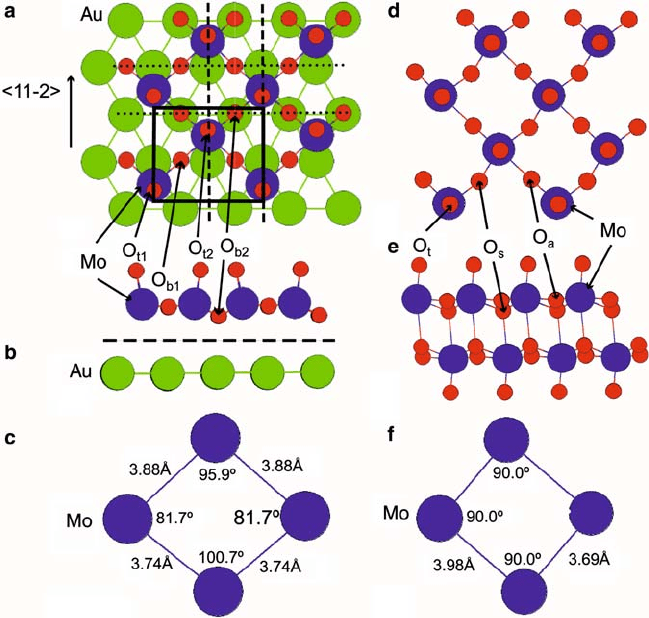
286 S.Y. Quek and E. Kaxiras
Fig. 9.3 (color online) Atomic structure of MoO
3
slabs. (a) Top view of MoO
3
monolayer on Au.
(b) Side view of MoO
3
monolayer on Au. (c) Top view of Mo sublattice in MoO
3
monolayer on
Au. (d) Top view showing half a bulk MoO
3
bilayer. (e) Side view of a bulk MoO
3
bilayer. (f) Top
view of Mo sublattice in half a bulk MoO
3
bilayer. Mo, Au, and O atoms are shown as large (blue),
medium (green), and small (red) spheres. MoO
3
units are close-packed along the diagonal of the
c.4 2/ unit cell, indicated by the black box. Dashed and dotted lines in (a) denote planes of
reflection and glide symmetry in the oxide monolayer, respectively. The view in (b) is that down
the glide planes, and shows MoO
3
units tilting backward and forward along the axes of reflection.
O
b1
and O
b2
denote the two inequivalent bridging O atoms in the unit cell of MoO
3
on Au, with
O
b2
nearer to the Au surface than O
b1
. The terminal O atoms in the MoO
3
monolayer on Au are
labeled O
t1
and O
t2
.Thedashed line in (b) denotes the plane relevant for the plot in Fig. 9.6.In
bulk MoO
3
, there are three distinct oxygen species, labeled O
t
(terminal), O
s
(symmetric bridging),
and O
a
(asymmetric bridging). Calculated bond lengths in the bulk are within 1–3% of experiment
(see Table 9.1).
the notation in Fig. 9.3,O
b2
is situated directly above an Au atom, whereas O
b1
is
above an Au bridge site. Mo sits in a threefold site, off-centered away from the Au
atoms below O
b2
.
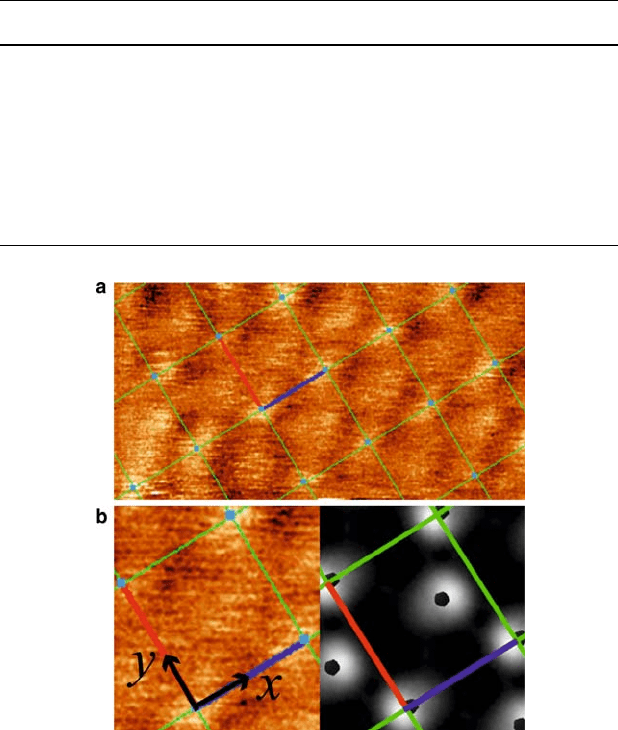
We performed simulations of the STM images expected for this system based
on the Tersoff–Hamann theory [34]. The bright spots in the STM images are found
9 Applications of Thin Film Oxides in Catalysis 287
Table 9.1 Bond lengths in bulk MoO
3
andintheMoO
3
monolayer on Au
Oxygen type
Number of
Mo neighbors
Calculated Mo–O bond lengths in
˚
A
(experimental values in brackets)
Bulk MoO
3
O
t
1 1.70 (1.67)
O
s
3 1.97 (1.95), 1.97 (1.95), 2.40 (2.33)
O
a
2 1.77 (1.73), 2.22 (2.25)
MoO
3
monolayer on Au
O
t1
11.69
O
t2
11.69
O
b1
2 1.95, 1.95
O
b2
2 1.98, 1.98
Fig. 9.4 (color online) STM images of the interior of the MoO
3
islands. (a) Experimental STM
image, collected at room temperature. (b) Left: Close-up section of experimental STM image.
Right: STM simulation corresponding to a tip-sample separation of 1.4
˚
A. The bias voltages were
0:580 V in both the experiment and the simulation. The tunneling current in the experiment
was 25.4 pA. A brighter color represents a more intense current. The lines show the c.4 2/
unit cells, which are 5:76
˚
A 4:99
˚
A. The bright spots in the experimental images are related
to lateral positions of terminal O atoms on the surface, which are marked by black pentagons in
the simulation. In each cell, there is a bright spot slightly off-center. In polar coordinates with
respect to the (x, y) axes, the off-center spot is at r D 4:3
˚
A;D 42
ı
in the simulation, and
r D 4:1 ˙0:4
˚
A;D 43 ˙ 4
ı
in the experiment.
corresponding to lateral positions of terminal O. Within the limits of experimental
variance, the relative positions of these spots are the same in theory and experiment
(Fig. 9.4), thus lending strong evidence to the predicted tilting of MoO
3
units.
288 S.Y. Quek and E. Kaxiras
Phonon frequencies of the MoO
3
monolayer were computed at the Brillouin zone
center, using the harmonic approximation. We found only 6 electron energy loss
spectroscopy (EELS)-active [35] phonon modes out of 24 possible ones. The cal-
culated frequencies, with corresponding experimental values in parentheses, are, in
cm
1
: 1030, 1020 (990), 804 (850), 430 (480), 351 (280), and 160 (not observed).
Noting that instrument resolution is about 80 cm
1
,andthat160 cm
1
is out of the
detection range, theoretical and experimental frequencies correspond fairly well,
especially since finite-size effects were neglected in the simulation. This correspon-
dence provides further evidence for the predicted symmetry properties.
The preceding results confirm unequivocally that the optimized structure matches
the experimental structure of the interior of the MoO
3
monolayer islands on
Au(111), without including defects. It is remarkable that the Mo sublattice from
the bulk monolayer distorts by as much as 11
ı
to fit the Au lattice (Fig. 9.3c).
Geometrical considerations indicate that the c.4 2/ unit cell is in fact the smallest
unit cell for which epitaxy can be achieved, if sufficient bonding between Mo atoms
through the bridging O bonds is to be preserved. The symmetry properties of the
monolayer are also dictated by the symmetries of the Au substrate – the reflection
symmetry in the oxide is matched by a reflection symmetry in the Au lattice, and the
glide plane symmetry in the oxide corresponds to a similar symmetry in the top Au
layer, if its relation to underlying Au layers is ignored. This flexibility of the oxide
lattice is achieved by the ability of the Mo–O bonds to rotate about one another: the
dihedral angles involving terminal oxygen atoms in the bulk monolayer are 0
ı
and
37
ı
(angle O
t
–Mo–O
s
–Mo in Fig. 9.3d), whereas the corresponding dihedral angles
in the relaxed MoO
3
monolayer on Au are 7–8
ı
.
Unlike bulk MoO
3
that has a bilayer structure and is semiconducting, the MoO
3
monolayer on the Au surface is semimetallic, as deduced from the density of states
(DOS) of the MoO
3
=Au system, projected onto the oxide slab (Fig. 9.5). The MoO
3
monolayer alone has a similar, semimetallic DOS. However, if this monolayer is
allowed to relax in the same supercell without the Au substrate, rows of Mo atoms
relax alternately toward rows of O
b1
and O
b2
, breaking the glide-plane symmetry,
and the monolayer becomes semiconducting. Analysis of the DOS of the semimetal-
lic MoO
3
monolayer reveals that Fermi level states are localized in the plane of Mo
and bridging O. These symmetry-degenerate states are split by a Jahn-Teller distor-
tion that leads to a semimetal-to-insulator transition with Mo relaxing toward a pair
of bridging O atoms to form stronger bonds.
We expect this oxide monolayer to exhibit interesting surface chemistry because
of the relative ease of promoting electrons across the Fermi level in a semimetal.
Indeed, H is found to adsorb more strongly than on bulk MoO
3
: the binding energies
for H at saturation coverage are, in eV, 3:39, 2:77,and3:13 on O
t
; O
s
,andO
a
,
respectively, in bulk MoO
3
[26], and 3:55, 3:95,and3:94 on the terminal O,
O
b1
,andO
b2
, respectively, in the MoO
3
monolayer on Au. In contrast to bulk MoO
3
,
the bridging oxygen atoms are more stable binding sites for H than the terminal
oxygen atoms. This is consistent with the localization of Fermi level states along
the plane of bridging O atoms. The adsorption of H on bridging O also provides
greater strain relief by breaking up the strained lattice.
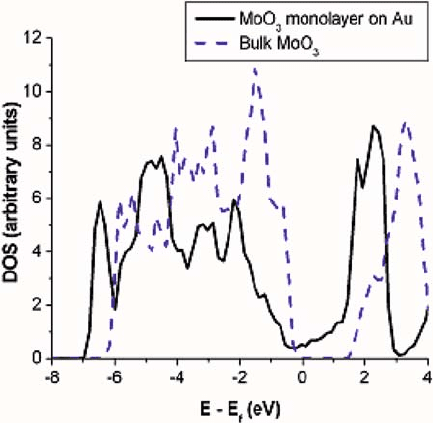
9 Applications of Thin Film Oxides in Catalysis 289
Fig. 9.5 Projected density of states (DOS) for the MoO
3
monolayer on Au and for bulk MoO
3
.
The above analysis suggests that the semimetallic character of the MoO
3
monolayer on Au can be attributed to the strained Mo–O bridging bonds. The
difference in energy between the relaxed and strained MoO
3
monolayers was
0.15 eV/supercell. The cohesive energy for the MoO
3
=Au system, with respect to
a relaxed unreconstructed Au(111) surface and the strained MoO
3
monolayer, was
0:24 eV/supercell. The energy cost of straining the MoO
3
monolayer is therefore
overcome by the gain in cohesive energy upon formation of the MoO
3
=Au interface.
To elucidate the nature of the MoO
3
=Au interaction, we plot the difference be-
tween the charge density of the MoO
3
=Au system, and the sum of charge densities of
isolated MoO
3
and Au slabs, frozen in configuration from the joint system (Fig. 9.6).
This charge-density difference reveals that the MoO
3
monolayer induces a redistri-
bution of electronic charge above the Au surface. The positively-charged Mo ions
draw electron density to the region directly underneath them. Each of these elec-
tron clouds is in turn attracted by the nearest Au atom since Au surface atoms are
electron-deficient. In this way, Mo is drawn closer to the Au atom nearest to it. At
the same time, the partial negative charges on O
b2
cause them to be attracted to Au
atoms directly beneath them. These interactions together cause the Mo–O
b2
bridging
bonds to strain resulting in semimetallic character. The electronic charge redistribu-
tion satisfies local bonding requirements, which allow the Au surface to act as the
other half of the MoO
3
bilayer, thereby stabilizing the monolayer nanocrystals. Each
Au surface atom is in turn bonded either to an O atom .O
b2
/ or a Mo atom, and as a
result, the surface reconstruction under the MoO
3
islands is lifted.
In situ STM studies suggest that the MoO
3
islands grow via aggregation of MoO
3
molecular species. Earlier theoretical work has shown that induced electrostatic

290 S.Y. Quek and E. Kaxiras
Fig. 9.6 Charge density difference between the MoO
3
=Au system and the sum of charge densities
of isolated MoO
3
and Au slabs, frozen in configuration from the joint system. The values are those
on a plane midway between MoO
3
and Au, as indicated by the dashed line in Fig. 9.3b. Half of the
Au atoms are hidden directly under O
b2
. Dark regions, corresponding to charge depletion, occur
below O
b2
,andbright regions, corresponding to charge accumulation, are seen between Mo and
the nearest Au atoms. Values of the charge density difference range from 0:0093 to 0:0133 e=
˚
A
3
.
interactions increase the cationic character of Mo as MoO
3
units build up to form
bulk MoO
3
[36]. Similarly, in our calculations, the local charge on Mo is larger in
the MoO
3
slab on Au than in a single MoO
3
molecule. The increased ionic char-
acter upon aggregation of MoO
3
molecular species allows the oxide to polarize the
electron gas at the MoO
3
=Au interface. Charge redistribution at the interface allows
the Au surface to serve as the other half of the MoO
3
bilayer, thus stabilizing the
monolayer structures, allowing nucleation and growth. The surface of these islands
corresponds to the natural cleavage plane of bulk MoO
3
and has a free energy of
only 0:05–0:07 J=m
2
[37]. In contrast, Au has a surface free energy of 1:62 J=m
2
[38]. Growth of the MoO
3
monolayer is thus driven by both a gain in interface
energy and a reduction in surface free energy.
Interestingly, the long straight edges of the ensuing islands (Fig. 9.2) run along
the h112idirections of Au, parallel to the herringbone pattern, and not the h312i
directions, diagonal to the c.4 2/ unit cell, along which MoO
3
units are close-
packed (Fig. 9.3a). The herringbone pattern is aligned parallel to straight island
edges, but tends to form sharp bends at rough island edges. The herringbone pattern
has the property of soliton-waves [39], therefore, absence of Au reconstruction be-
neath the islands imposes hard-wall boundary conditions on these waves, causing
the herringbone pattern to become locally parallel to the island edges. The distinct
correlation between straight island edges and the herringbone direction points to-
ward an interplay between the herringbone structure and the MoO
3
islands that
affects the overall pattern developed on the surface. Further theoretical and experi-
mental investigation of kinetic effects will substantially clarify the picture.
9 Applications of Thin Film Oxides in Catalysis 291
9.2.4 Concluding Remarks
In this section, we have demonstrated that while MoO
3
exists as bilayers in the bulk
crystal, MoO
3
monolayer nanocrystals can be grown on the Au(111) surface. These
structures are also distinctly different from previously reported ramified MoO
3
is-
lands grown on Au(111) [40]. The observed flexibility of dihedral bond angles is
likely to be common to many transition metal oxides, especially those with more
than one structural phase in the bulk. In fact, it is likely that the growth mechanism
proposed herein is general enough so that novel structures of such oxides can be
grown on metal surfaces by condensing molecular species, which become increas-
ingly ionic, interacting with the substrate to create a wetting oxide layer. Thin films
of some of these oxides have been grown on metal surfaces [14, 41]. For exam-
ple, novel V
x
O
y
structures were recently grown on Pd(111) and understood by first
principles energetic arguments [41]. Our analysis shows explicitly how the metal-
lic substrate can induce strain in an oxide monolayer, resulting in changes in the
electronic properties of the oxide, thereby leading to interesting surface chemistry.
These results suggest that the metallic substrate may be used as a handle to tune
the electronic properties of interface-mediated oxide structures. The ability to grow
crystalline oxide structures epitaxially on metal surfaces thus provides a first step
toward synthesizing oxide systems with controllable properties.
9.3 Ultrathin Titania Films as Active Supports for Supported
Au Catalysts
9.3.1 Background and Introduction
The role of oxide supports in promoting catalytic activity of metal catalysts has
been extensively discussed in the literature. Much of this discussion has focused on
the availability of active sites at the metal/oxide interface [42,43]. In some cases, the
metal catalyst is predicted to form a surface oxide at this interface, further promot-
ing catalysis [44]. The catalytic activity at interface sites has also been explained in
terms of hot electron flow induced by exothermic reactions such as carbon monox-
ide oxidation [7]. In addition, the oxide support can enhance the catalytic activity
of the metal nanostructure by altering its electronic properties prior to catalysis,
via charge transfer [20, 45]andstrain[46, 47]. Electron transfer from the oxide
support to the metal catalyst has been linked to the so-called strong metal support
interaction [48].
An oxide/metal system that has attracted tremendous attention is that of oxide-
supported Au nanoparticles and films, which act as excellent catalysts [49]. Theo-
retical studies indicate that the active sites include under-coordinatedAu atoms [50]
with rough orbitals [51], and sites at the Au/oxide interface [42–44]. Experiments
also suggest that the activity of titania-supported Au films increases markedly when
292 S.Y. Quek and E. Kaxiras
the Au thickness is reduced to one nearest neighbor distance in bulk Au (so-called
“bilayers”) [9]. A key insight from theoretical studies was that the ability of Au
atoms in a nanoparticle to rearrange in response to adsorbates is essential for O
2
adsorption [52]; this effect was called “fluxionality” of the nanoparticle. Here, we
show that the notion of fluxionality can be extended to the Au/oxide interface for
ultrathin Au films on ultrathin reduced titania.
In contrast to bulk oxide supports, ultrathin oxide layers can exhibit special be-
havior by enabling the coupling of structural distortions and charge transfer beyond
that allowed in the bulk. In this section, we propose that this behavior results in a
“dynamic” active role of the ultrathin oxide support in promoting catalysis of sup-
ported metal catalysts [53]. In particular, when the support is an ultrathin reducible
oxide film, atoms at the buried metal/oxide interface can rearrange in response to
the presence of adsorbates on the metal film, provided the latter is sufficiently thin.
This atomic relaxation at the interface lowers the total energy of the system, thereby
stabilizing adsorption. We call the ability of interfacial atoms to rearrange during
adsorption “dynamic interface fluxionality.” We demonstrate this effect on a model
structure consisting of a thin Au film on an ultrathin titania layer supported on a
molybdenum slab, and suggest that it is of more general nature. Specifically, we
expect that when the metal film forms strong covalent bonds with the reducible ul-
trathin oxide layer, whereas the latter does not interact strongly with its support to
render it a rigid structure, dynamic interface fluxionality can take place. This effect
may be exploited to design better catalysts and sensors by replacing traditional re-
ducible oxide supports with ultrathin oxide films. Recent advances in the control of
ultrathin film growth [13,14] indicate that this is a practical possibility.
9.3.2 Motivation
The possibility of wetting an ultrathin titania support with Au films was recently
demonstrated, with Mo(112) as a substrate on which the TiO
2
thin film was grown
[9]. CO oxidation activity in this system was more than 45 times greater than that
reported for other Au/titania catalysts. The atomic structure of this system is un-
known. However, two salient features are the strong interaction between Au and
titania through Au–Ti bond formation and the presence of ultrathin reduced titania
beneath the Au film. Both of these effects have precedent in other systems [54,55].
Reducible oxides grown on a metal substrate have lower oxidation states than bulk
phases due to the reducing character of the metal surface (in the present case, Mo)
[54]. On bulk TiO
2
surfaces, Au binds almost exclusively to reduced Ti sites [55].
The availability of such sites throughout the ultrathin titania film thus allows wetting
by Au. What is not clear is how a strong interaction with buried ultrathin titania, or
a small Au thickness, can enhance the activity of Au/titania catalysts [56].
Motivated by these questions, and knowledge that the CO oxidation rate is lim-
ited by the availability of O
2
or adsorbed O on the catalyst [42, 57], we study O
2
and O adsorption in a model ultrathin Au/titania system.