Pecharsky V.K., Zavalij P.Y. Fundamentals of Powder Diffraction and Structural Characterization of Materials
Подождите немного. Документ загружается.


5
92
Chapter
6
J. Ruis, Solution of Patterson-type syntheses with the direct methods sum
function, in: Structure determination from powder diffraction data. IUCr
monographs on crystallography 13.
W.
I.
F.
David,
K.
Shankland, L.B.
McCusker, and Ch. Baerlocher, Eds., Oxford University Press, Oxford,
New York (2002).
K. Shankland and
W.
I.
F.
David, Global optimization strategies, in:
Structure determination from powder diffraction data. IUCr monographs
on crystallography 13.
W.
I.
F.
David, K. Shankland, L.B. McCusker,
and Ch. Baerlocher, Eds., Oxford University Press, Oxford, New York
(2002).
P.
G.
Bruce and
Y.
G.
Andreev, Solution of flexible molecular structures
by simulated annealing, in: Structure determination fi-om powder
diffraction data. IUCr monographs on crystallography 13.
W.
I.
F.
David,
K. Shankland, L.B. McCusker, and Ch. Baerlocher, Eds., Oxford
University Press, Oxford, New York (2002).
10. L. B. McCusker and Ch. Baerlocher, Chemical information and intuition
in solving crystal structures. NCr monographs on crystallography 13.
W.
I.
F.
David, K. Shankland, L.B. McCusker, and Ch. Baerlocher, Eds.,
Oxford University Press, Oxford, New York (2002).
11.
D.
Louer, Advances in powder diffraction analysis, Acta Cryst. A54, 922
(1998).
12. D.M. Poojary and A. Clearfield, Application of x-ray powder diffraction
techniques to the solution of unknown crystal structures, Acc. Chem.
Res. 30,414 (1997).
13.K.D.M. Harris and M. Tremayne, Crystal structure determination from
powder diffraction data, Chem. Mater. 8, 2554 (1996).
14. C. Giacovazzo, Direct methods and powder data: State of the art and
perspectives, Acta
Cryst. A52,
33
1 (1996).
15. J.A. Kaduk, Use of the Inorganic Crystal Structure Database as a problem
solving tool, Acta Cryst. B58, 370 (2002).
16. A. Le Bail, SDPD
-
Structure Determination from Powder Diffraction
-
Database of bibliography and methods, http://sdpd.univ-
lemans. fr/iniref.html.

Crystal structure solution
593
17.
V.
Fane-Nicolin and
R.
Cern);,
FOX,
"Free objects for crystallography":
a modular approach to
ab
initio
structure determination from powder
diffraction,
J.
Appl. Cryst.
35,
734
(2002).

5 94 Chapter
6
6.20
Problems
Answers to all problems listed below are located in the file Chapter-6-
Problems-Solutions.vdf on the CD accompanying this book.
1. The compound Mn5Si3012 crystallizes in the space group Ia3d with lattice
parameter a
=
11.85 A. The measured gravimetric density,
p
=
4.4 g/cm3.
Calculate the number of formula units in the unit cell and the number of
atoms of each kind. Make a suggestion, which sites can be occupied by
the different types of atoms in this unit cell.
2. The compound Co2Mn308 crystallizes in the space group Prn1-12~ with
lattice parameters a
=
5.743,
b
=
4.915 and
c
=
9.361 A. Assuming a
reasonable density of a 3d-metal oxide (3 to 6 g/cm3), find the number of
formula units in the unit cell and calculate the x-ray density of the
material.
3. Cobalt oxide, COO, crystallizes in the cubic crystal system, space group
Fm3m, a
=
4.26 A. The measured gravimetric density of the oxide is
p
=
6.438 g/cm3. Using only these data, solve its crystal structure (find
positions of atoms that make chemical and physical sense and have
reasonable interatomic distances).
4.
The compound TaMn203 crystallizes in the hexagonal crystal system and
belongs to the space group P61mmm with
a
=
5.321,
c
=
3.578
A.
The
measured gravimetric density of the material is
p
=
6.30 g/cm3. Using
only these data, solve the crystal structure of the material (find positions
of atoms that make chemical and physical sense and have reasonable
interatomic distances).
5. Hexamethylenetetramine molecule, C6H12N4 (hmta), has the configuration
of a tetrahedron, where the comers are occupied by nitrogen atoms,
which are bonded with each other by means of six methylene, CH2,
groups located above the mid points of the edges of the tetrahedron as
shown in
Figure
6.44.
The compound crystallizes in the cubic crystal
system, space group I43m, a
=
7.05 A. The measured gravimetric
density is
p
=
1.33 g/cm3. Assuming that the tetrahedron is ideal and that
the C-N distances are 1.49 A, solve this crystal structure (i.e. determine
the coordinates of non-hydrogen atoms) using data provided above
including both the symmetry of the lattice and hmta molecule. To
simplify calculations, consider the following: the distance fkom the center
(X)
of the tetrahedron to the C atoms is
6x-c
=
1.72 A, and to the N
atoms,
6x,
=
1.49 A.
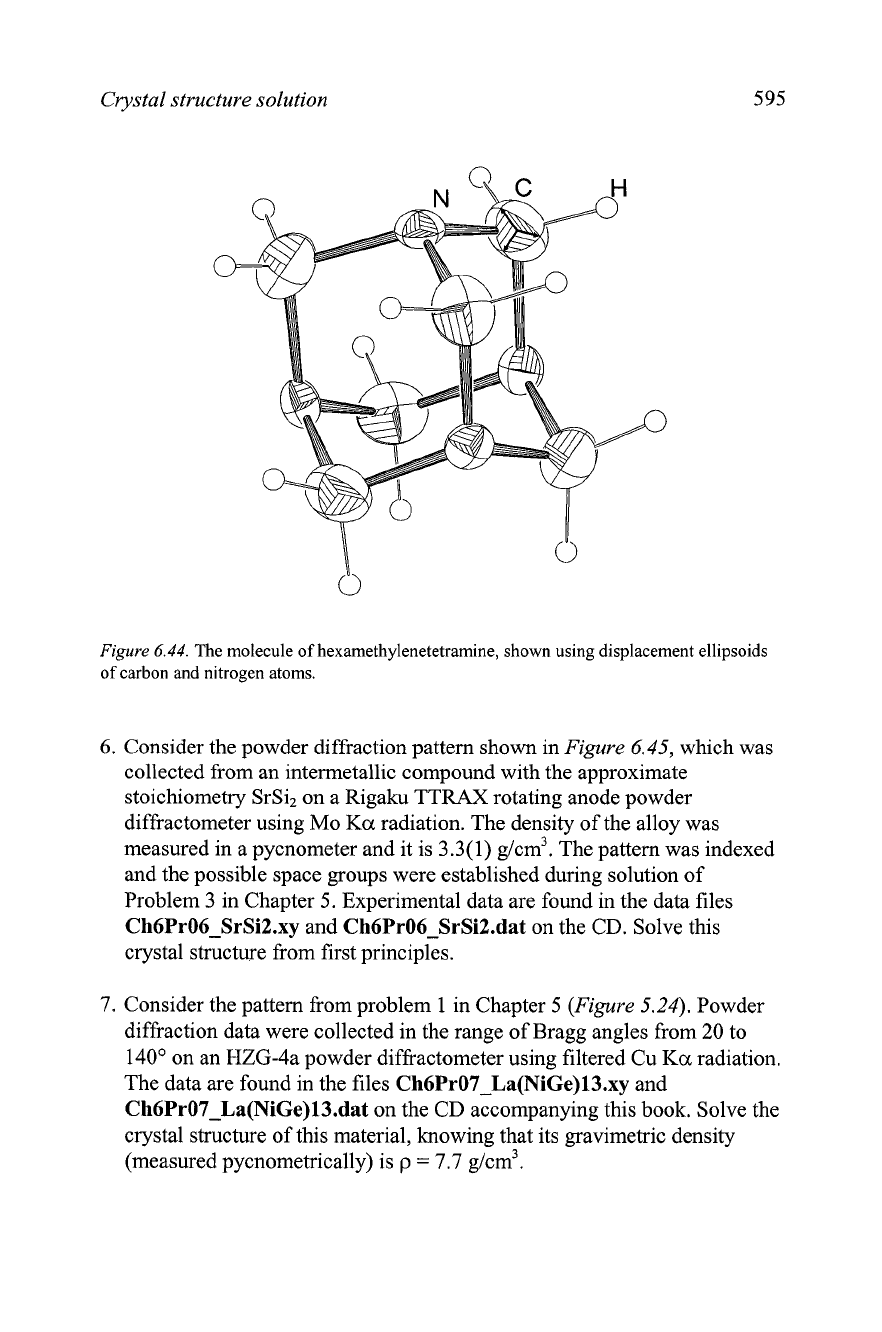
Crystal structure solution
Figure
6.44.
The molecule of hexamethylenetetramine, shown using displacement ellipsoids
of carbon and nitrogen atoms.
6.
Consider the powder diffraction pattern shown in Figure
6.45,
which was
collected from an intermetallic compound with the approximate
stoichiometry SrSi2 on a Rigaku TTRAX rotating anode powder
diffractometer using Mo
Ka
radiation. The density of the alloy was
measured in a pycnometer and it is 3.3(1) g/cm3. The pattern was indexed
and the possible space groups were established during solution of
Problem 3 in Chapter
5.
Experimental data are found in the data files
Ch6Pr06-SrSi2.x~
and
Ch6Pr06-SrSi2.dat
on the CD. Solve this
crystal structure from first principles.
7.
Consider the pattern from problem 1 in Chapter
5
(Figure
5.24).
Powder
diffraction data were collected in the range of Bragg angles from 20 to
140' on an HZG-4a powder diffractometer using filtered Cu
Ka
radiation.
The data are found in the files
Ch6Pr07-La(NiGe)l3.xy
and
Ch6Pr07_La(NiGe)l3,dat
on the CD accompanying this book. Solve the
crystal structure of this material, knowing that its gravimetric density
(measured pycnometrically) is
p
=
7.7
g/cm3.
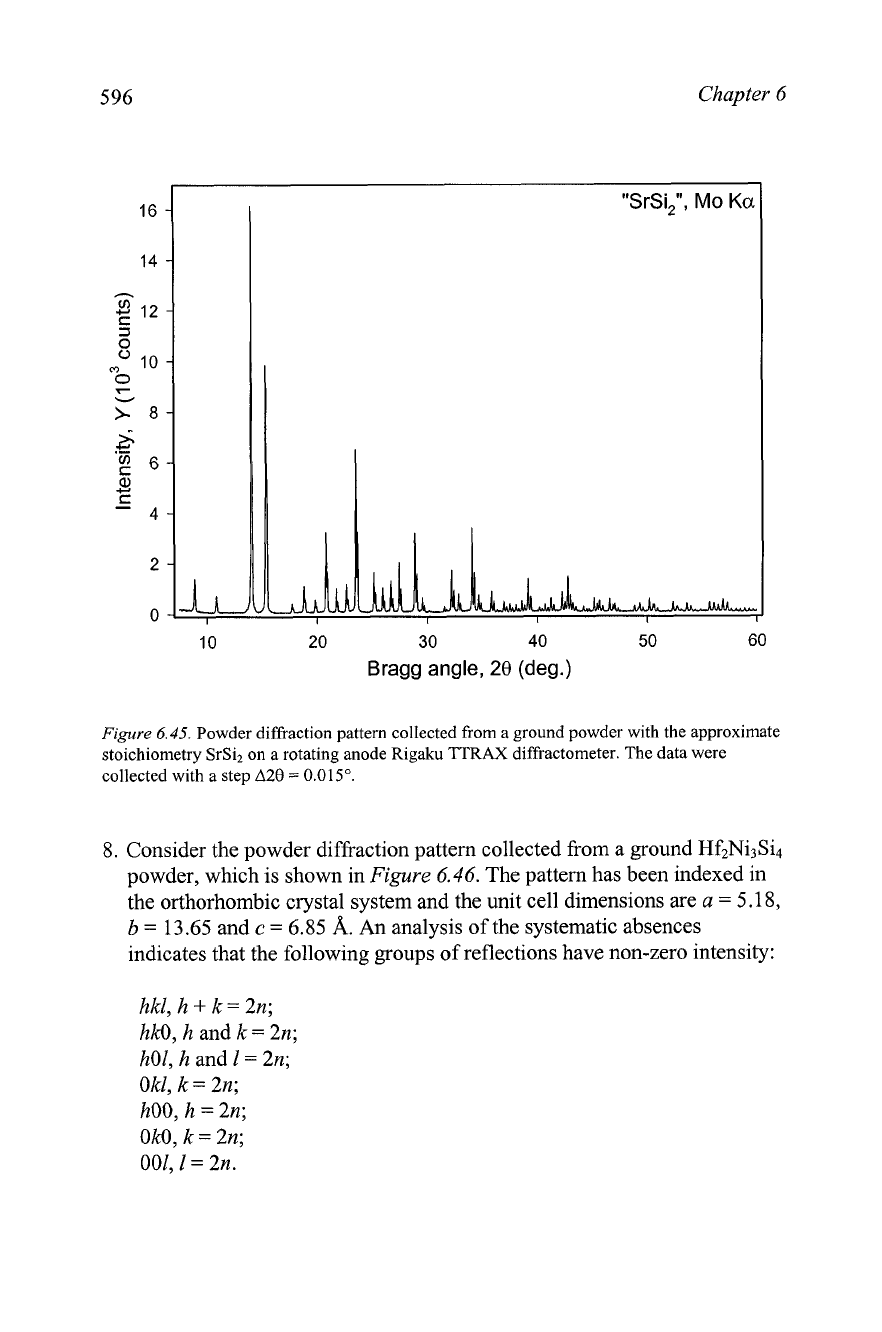
Chapter
6
30
40
50
60
Bragg angle,
28
(deg.)
Figure
6.45.
Powder diffraction pattern collected from a ground powder with the approximate
stoichiometry SrSiz on a rotating anode Rigaku TTRAX diffractometer. The data were
collected with a step
A28
=
0.015".
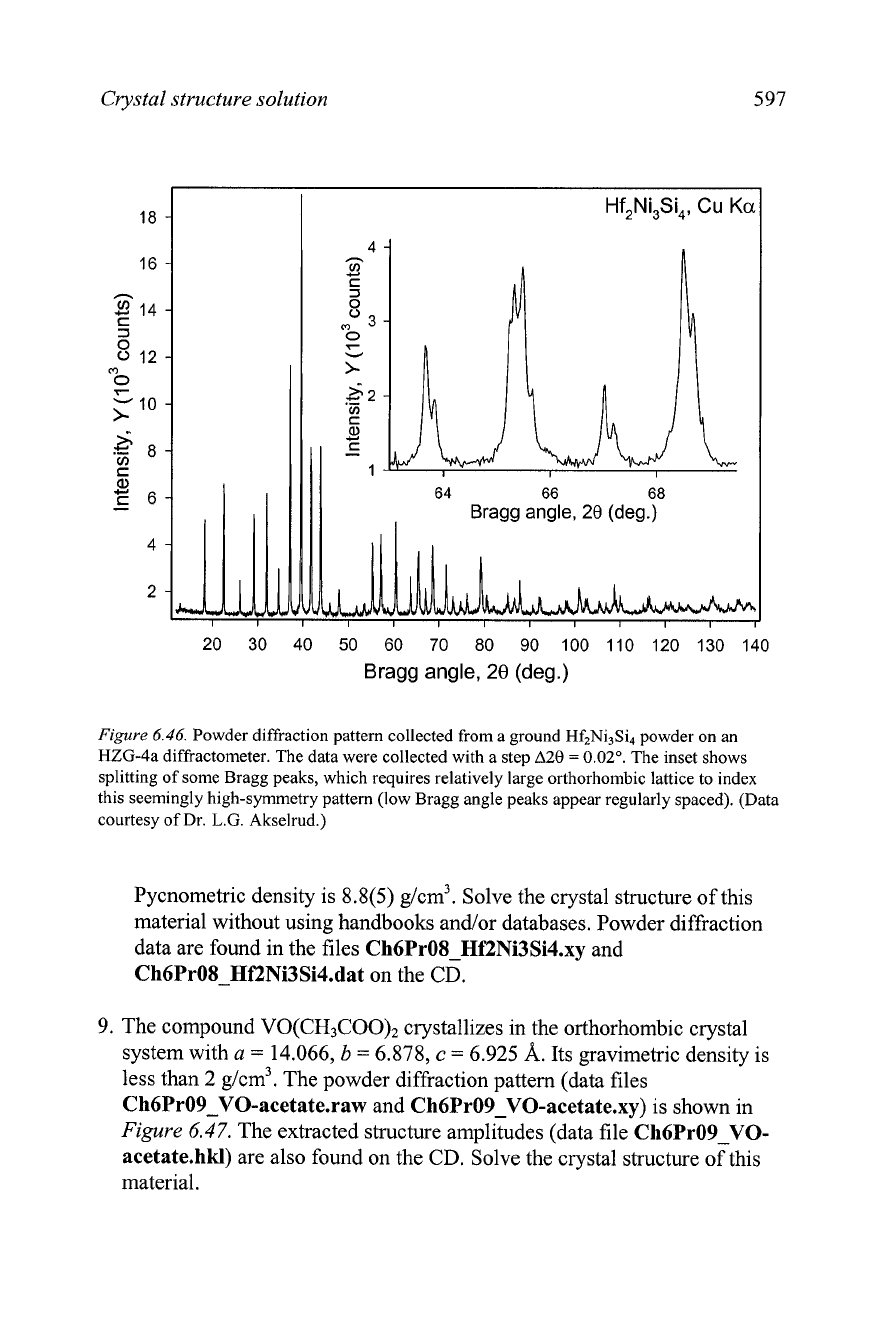
8.
Consider the powder diffraction pattern collected from a ground Hf2Ni3Si4
powder, which is shown in
Figure 6.46.
The pattern has been indexed in
the orthorhombic crystal system and the unit cell dimensions are
a
=
5.18,
b
=
13.65
and
c
=
6.85
A.
An
analysis of the systematic absences
indicates that the following groups of reflections have non-zero intensity:
hkl, h
+
k
=
2n;
hkQ, h
and
k
=
2n;
h01, h
and
1
=
2n;
Okl,
k
=
2n;
hOO, h
=
2n;
OH,
k
=
2n;
001,1= 2n.
Crystal structure solution
597
II
I
64
66
68
Bragg
angle,
28
(deg.)
20 30 40 50 60 70 80 90 100 110 120 130 140
Bragg angle,
28
(deg.)
Figure
6.46.
Powder diffraction pattern collected from a ground Hf2Ni3Si4 powder on an
HZG-4a diffractometer. The data were collected with a step
A28
=
0.02'.
The inset shows
splitting of some Bragg peaks, which requires relatively large orthorhombic lattice to index
this seemingly high-symmetry pattern (low Bragg angle peaks appear regularly spaced). (Data
courtesy of Dr.
L.G.
Akselrud.)
Pycnometric density is 8.8(5) g/cm3. Solve the crystal structure of this
material without using handbooks and/or databases. Powder diffraction
data are found in the files Ch6PrOSHf2Ni3Si4.xy and
Ch6PrO8-HEZNi3Si4.dat on the CD.
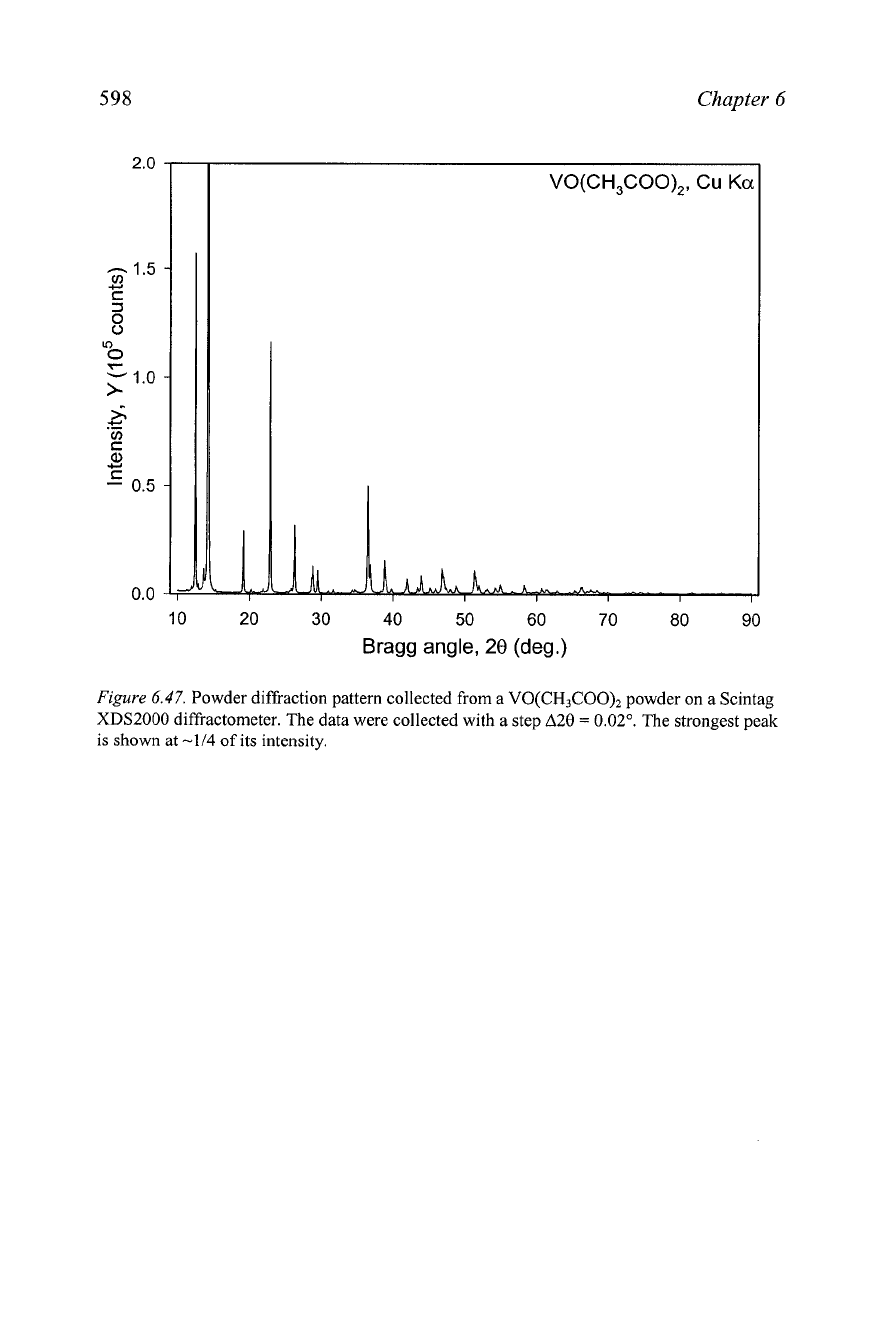
9. The compound VO(CH3C00)2 crystallizes in the orthorhombic crystal
system with
a
=
14.066,
b
=
6.878,
c
=
6.925
A.
Its gravimetric density is
less than 2 g/cm3. The powder diffraction pattern (data files
Ch6Pr09-VO-acetate.raw and Ch6Pr09-VO-acetate-xy) is shown in
Figure
6.47.
The extracted structure amplitudes (data file Ch6Pr09-VO-
acetate.hk1) are also found on the CD. Solve the crystal structure of this
material.
Chapter
6
10 20 30 40 50 60 70 80 90
Bragg angle,
20
(deg.)
Figure
6.47.
Powder diffraction pattern collected from
a
VO(CH3C00)2
powder on a Scintag
XDS2000 diffractometer. The data were collected with a step A20
=
0.02".
The strongest peak
is shown at
-114
of its intensity.

Chapter
7
CRYSTAL STRUCTURE REFINEMENT
7.1
Introduction
As briefly mentioned in the previous chapter, the determination of a
crystal structure may be considered complete only when multiple pattern
variables and crystallographic parameters of a model have been fully refined
against the observed powder diffraction data. Obviously, the refined model
should remain reasonable from both physical and chemical standpoints. The
refinement technique, most commonly employed today, is based on the idea
suggested in the middle 1960's by
Riet~e1d.I'~ The essence of Rietveld's
approach is that experimental powder diffraction data are utilized without
extraction of the individual integrated intensities or the individual structure
factors, and all structural and instrumental parameters are refined by fitting a
calculated profile to the observed data.
To a certain extent, the Rietveld method (also known as the full pattern or
the full profile refinement)
is
similar to the full pattern decomposition using
Pawley and/or Le Bail algorithms, except that the values of the integrated
'
Hugo M. Rietveld (b. 1932). Dutch physicist, who between 1964 and 1966 demonstrated
that accurate determination of crystal and magnetic structures, is possible using neutron
diffraction data from powders. His approach was later extended to x-rays. At present,
crystal structures of hundreds, if not thousands of polycrystalline materials, are studied and
refined every year using the Rietveld method. In recognition of the "Rietveld method", the
Royal Swedish Academy of Sciences awarded the author, Dr. Hugo M. Rietveld, the
Aminoff prize in 1995. For more information, see
http:/ihome.wxs.nl/-rietv0251
or the
mirror site at
http:Nccpl4.sims.nrc.ca/ccp/web-mirrors/hugorietveld/-rietvO25/index.html.
The following two papers are considered seminal: H.M. Rietveld, Line profiles of neutron
powder-diffraction peaks for structure refinement, Acta Cryst.
22,
151 (1967); H.M.
Rietveld, A profile refinement method for nuclear and magnetic structures,
J.
Appl. Cryst.
2,
65 (1969).

600
Chapter
7
intensities are no longer treated as free least squares variables (Pawley) or
determined iteratively after each refinement cycle (Le Bail).' They are
included into all calculations as functions of relevant geometrical, specimen
and structural parameters (see sections 2.10 and 2.11).
Full profile refinement is computationally intense and employs the non-
linear least squares method (section
6.6), which requires a reasonable initial
approximation of many free variables. These usually include peak shape
parameters, unit cell dimensions and coordinates of all atoms in the model of
the crystal structure. Other unknowns (e.g, constant background, scale
factor, overall atomic displacement parameter, etc.) may be simply guessed
at the beginning and then effectively refined, as the least squares fit
converges to a global minimum.
When either Le Bail's or Pawley's
techniques were employed to perform a full pattern decomposition prior to
Rietveld refinement, it only makes sense to use suitably determined relevant
parameters (background, peak shape, zero shift or sample displacement, and
unit cell dimensions) as the initial approximation.
The successful practical use of the Rietveld method, though directly
related to the quality of powder diffi-action data (the higher the quality, the
better the outcome), largely depends on the experience and the ability of the
user to properly select a sequence in which various groups of parameters are
refined. Regardless of the relatively long history of the method, it is certainly
true that almost everyone familiar with the technique has
hislher own set of
"unique" secrets about how to make the refinement stable, complete and
triumphant. Therefore, in this chapter we will simply introduce the basic
theory of Rietveld's approach, followed by a series of hands-on examples to
demonstrate the refinement of crystal structures with various degrees of
completeness and complexity. Every example considered in this book is
supplemented by actual experimental data given on the
CD,
thus allowing
the reader many opportunities to follow our reasoning, as well as to create
and test hisker own strategies leading to the successful completion of the
crystal structure determination
from powder
diffraction
data.
Just as in the case of full pattern decomposition, we will use two fi-eely
available software codes (LHPM-Rietica2 and
GSAS3)
to carry out Rietveld
refinements using either or both x-ray and neutron diffraction data. Many
'
We introduce this analogy for clarity, even though both Pawley's and Le Bail's techniques
were developed following Rietveld's work.
LHPM-Rietica (authors: B.A. Hunter and
C.J.
Howard) may be downloaded from
ftp://ftp.ansto.gov.au/pub/physics/neutron/rietveld/RieticaLHPM95/
or
via
a link at
http://www.ccp14.ac.uk.
GSAS (authors A.C. Larsen and R. Von Dreele) may be downloaded from
http://public.lanl.gov/gsas/
or
via
a link at http://www.ccpl4.ac.uk. A convenient graphic
user interface for GSAS (author: B.H. Toby) may be downloaded from
http://www.ncnr.nist.gov/programs/crystallography/ or
via
http://www.ccpl4.ac.uk.

Crystal structure refinement
60
1
more software products, including Fullprof, Rietan, XRS-82 and others may
be downloaded from the International Union of Crystallography andlor
Collaborative Computational Project 14 Web sites1 When available, readers
may also employ a variety of commercial crystallographic software suites of
pr~grams.~
It is worth noting that parameters identical to those listed in our examples
can be expected only when the same computer codes are used to perform full
profile refinement due to small but detectable differences in the
implementation of the Rietveld method by various software developers.
Furthermore, even when the same version of an identical computer program
is employed to treat the same set of experimental data, small deviations may
occur due to subjective decisions, such as when to terminate the refinement.
In
the latter case, however, the differences should be within a few least
squares standard deviations.
7.2
The Rietveld method
Consider
Figure
7.1,
which shows both the observed and calculated
powder diffraction patterns of LaNi4,ssSno.ls for a narrow range of Bragg
angles. Assuming that
-
we have an adequate structural model (see Chapter
6),
which makes both
physical and chemical sense;
-
the model yields correct (i.e. close to experimentally observed) integrated
intensities (see Chapter 2), and
-
we have suitable peak shape and background functions (see Chapters 2
and 4, respectively),
the fully refined crystal structure of a material should result in a calculated
powder diffraction pattern closely resembling collected data.
In
other words,
the difference between the measured and calculated powder diffraction
profiles should be close to zero. This basic idea, extended to the entire
powder diffraction pattern, is the foundation of the Rietveld method.
The development of the Rietveld method and especially subsequent work
that showed its applicability to processing conventional x-ray powder
diffraction
data,3 began a remarkable era, which continues today, where
'
IUCr: http://www.iucr.org; CCP-14: http://www.ccpl4.ac.uk.
Rietveld refinement programs are included in software products sold by Bruker
(http://ww.bruker-axs.com/production/indexnn.htm),
Philips
(http://www-us.analytica1.
philips.com/products/xrd/),
RigakuIUSA
(http://www.RigakuMSC.com/xrd/index.shtml),
STOE
&
Cie, Gmbh
(http:llww.stoe.com/productslindex.htm),
Reflex or Reflex+
modules in Materials Studio or Cerius2 suites from Accelrys Inc. (www.accelrys.com),
Jade and jPOWD from MDI, Materials Data Inc. (www.materialsdata.com) and others.
J.
Malmros and J.O. Thomas, Least-squares structure refinement based on profile analysis
of powder film intensity data measured on an automatic microdensitometer, J. Appl.
Cryst.