Moss Tom. DNA-protein interactions: principles and protocols
Подождите немного. Документ загружается.


Cleavage of DNA by Histone–Fe(II) EDTA 275
275
From:
Methods in Molecular Biology, vol. 148: DNA–Protein Interactions: Principles and Protocols, 2nd ed.
Edited by: T. Moss © Humana Press Inc., Totowa, NJ
19
Site-Directed Cleavage of DNA
by Linker Histone-Fe(II) EDTA Conjugates
David R. Chafin and Jeffrey J. Hayes
1. Introduction
The ordered and regular packaging of eukaryotic DNA within the chromatin
complex allows the efficient utilization of this substrate for nuclear processes
such as DNA replication, transcription, recombination, and repair (1,2). Thus,
an understanding of the organization of protein–DNA interactions and associa-
tions within the chromatin complex is a prerequisite for a complete molecular
description of these processes. For example, the linker histone protein plays
crucial roles in the stability and organization of the chromatin fiber (3,4). This
multidomain protein undoubtedly makes complicated and diverse but poorly
understood interactions within the chromatin fiber (1). Currently, there is dis-
agreement regarding the site of association of the globular domain of this pro-
tein within the nucleosome proper (5,6). Moreover, the molecular interactions
and chemical activities of its N- and C-terminal tails are most likely modulated
by the multiple posttranslational phosphorylation events known to occur within
these domains (1,2). Thus, the linker histone tails represent critical points for
signal transduction within the chromatin complex likely to be manifested as
structural alterations within chromatin.
To elucidate the multiple interactions between the linker histone protein and
several model chromatin complexes, we have opted for a site-directed chemi-
cal cleavage methodology originally introduced by Ebright and colleagues and
Fox and colleagues (7,8). This method relies on targeting a DNA cleavage
reagent via the unique nucleophilic properties of a cysteine sulfhydryl engi-
neered at rationally selected locations within the protein of interest. The linker
histone protein is a particularly suitable candidate for this type of approach
because only in one rare instance (9) has this protein been found to contain a
276 Chafin and Hayes
cysteine residue. The single sulfhydryl group within the protein is modified
with a bifunctional reagent that contains a cysteine-specific moiety at one end
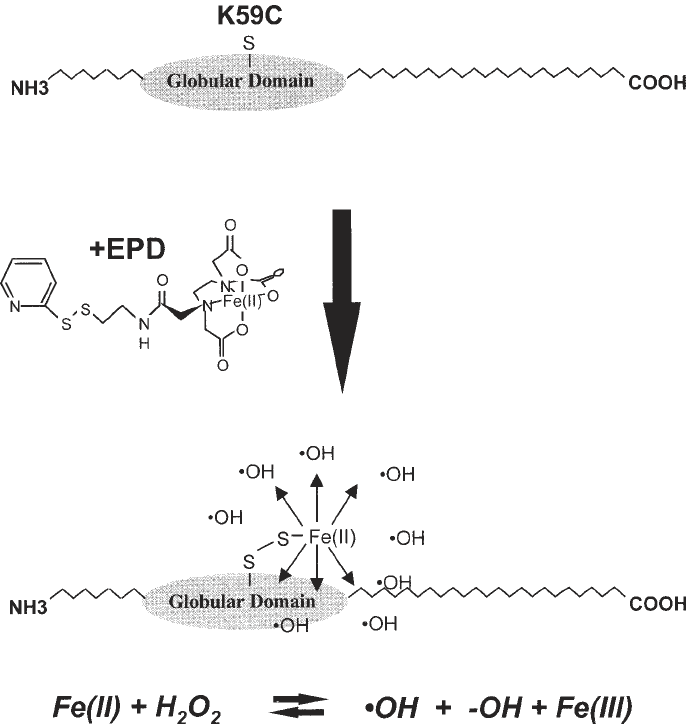
and an iron(II)-based DNA cleavage reagent at the other (Fig. 1) (7,8,10–12).
The protein is then assembled into the chromatin complex of interest and DNA
cleavage is initiated by standard Fenton chemistry (7,8). The DNA from such
complexes is prepared and the location of DNA cleavage is mapped to single-
base-pair resolution on DNA sequencing gels.
2. Materials
2.1. Construction of Cysteine Substituted Protein
2.1.1. Point Mutation by PCR
1. Oligonucleotide primers: Two oligonucleotide primers complimentary to the 5'
and 3' ends of the sequence to be amplified are needed. In addition, if the codon
to be altered is located more than approx 10–15 nucleotides from the end of the
coding sequence, one additional primer is needed that must contain the sequence
substitutions for the altered codon flanked by 12–15 nucleotides of complemen-
tary sequence on each side. Store at –20°C.
2. 10X stock containing all four dNTPs at 10 mM concentration each.
3. A source of clean reliable 18 meg Ω water, free of chemical contaminants.
4. 10X polymerase chain reaction (PCR) buffer; can be obtained commercially from
the supplier of the PCR enzyme of choice.
5. Vent or Taq DNA polymerase; can be obtained from commercial sources.
2.1.2. Ligation and Transformation of PCR Insert
into DH5
α
or BL21 Cells
1. DH5α or BL21 cells can be obtained commercially or prepared in competent
form; store at –70°C.
2. Luria–Bertani (LB) medium, sterile.
3. 1000X stock of ampicillin (100 mg/mL).
4. LB–agar plates containing 0.1 mg/mL ampicillin.
2.1.3. Overexpression and Purification of Mutant Histone Proteins
1. 100X (0.2 M) stock of isopropyl β-D-thiogalactopyranoside (IPTG).
2. Luria–Bertani (LB) medium, sterile.
3. 10 mg/mL lysozyme solution.
4. 1000X PMSF (phenylmethylsulfonyl fluoride): 100 mM in ethanol.
5. Triton-X100 detergent.
6. A 2-M solution of NaCl.
7. A 50% (v/v) slurry of Bio-Rex 70, 100-mesh chromatography resin (Bio-Rad).
8. 10 mM Tris-HCl, pH 8.0, and 1 mM EDTA solutions containing 0.5 M, 0.6 M, 1.0 M,
and 2.0 M NaCl.
Cleavage of DNA by Histone–Fe(II) EDTA 277
2.2. Reduction and Modification of Cysteine-Substituted Proteins
with EDTA-2-aminoethyl 2-pyridyl disulfide (EPD)
1. 1 M stock of DTT (dithiothreitol), made fresh; store at –20°C.
2. A 50% slurry of Bio-Rex 70, 100–200 mesh chromatography resin (Bio-Rad).
3. 10 mM Tris-HCl, pH 8.0 solutions containing 0.5 M, 0.6 M, 1.0 M, and 2.0 M NaCl.
Fig. 1. Lysine at position 59 within the globular domain of histone H1 was changed
to a cysteine residue (K59C, top). The free sulfhydryl group on the cysteine residue
was coupled to the DNA cleavage reagent EPD (middle). Hydroxyl radicals were
produced from the Fe
2+
center contained within the EPD moiety by standard Fenton
chemistry (bottom and equation).
278 Chafin and Hayes
4. 0.3 M stock of EPD synthesized according to refs. 7 and 8. Alternatively,
iodoacetamido-1,10 phenanthroline–Cu
2+
can be employed in place of EPD
(Molecular Probes, Eugene, OR).
5. Disposable 10 mL plastic chromatography columns (Bio-Rad).
6. Coomassie blue stain: 45% methanol, 10% acetic acid, and 0.25% coomassie
brilliant blue R250.
2.3. Radioactive End-Labeling
of a Purified DNA Restriction Fragment
1. Linear DNA fragment with convenient restriction sites on either end, previ-
ously phosphatased.
2. 10X T
4
polynucleotide kinase buffer (supplied with enzyme).
3. [γ-
32
P]dATP, 6000 Ci/mmol.
4. T
4
polynucleotide kinase 10,000 units/mL (Promega).
5. 2.5 M ammonium acetate.
6. 95% Ethanol, –20°C.
7. 70% Ethanol, –20°C.
8. 10% SDS stock solution.
9. Alkaline phosphatase from calf intestine (Boehringer Mannheim)
10. TE buffer: 10 mM Tris-HCl, pH 8.0, and 1 mM EDTA.
2.4. In Vitro Reconstitution of Nucleosomes
1. 10 mM Tris-HCl, pH 8.0, and 1 mM EDTA solutions containing 1.2 M, 1.0 M,
0.8 M, and 0.6 M NaCl.
2. TE buffer.
3. Stock of 6000–8000 molecular-weight cutoff dialysis tubing, 14 mm in diameter.
4. Stock of sonicated calf thymus (CT) DNA, approx 1–2 mg/mL.
5. Stock of 5 M NaCl.
6. Source of purified core histone proteins H2A/H2B and H3/H4, ours were puri-
fied from chicken erythrocytes (see Note 7).
2.5. Maxim–Gilbert G-Specific Reaction
1. 10X G-specific reaction buffer: 0.5 M NaCacodylate and 10 mM EDTA.
2. Dimethylsulfate (DMS) (Sigma).
3. G-reaction stop buffer: 1.5 M Na acetate, 1 M β-mercaptoethanol and 0.004 µg/µL
sonicated calf thymus DNA.
4. Piperidine (neat, 10 M stock) (Sigma).
2.6. Chemical Mapping of Protein–DNA Interactions with EPD
1. 0.7% agarose made with 0.5X TBE. (Note: Treat all solutions with chelex 100 resin
[Bio-Rad] to remove adventitious redox-active metals.)
2. Histone dilution buffer: 10 mM Tris-HCl, pH 8.0, and 50 mM NaCl.
3. Stock solution of 20 mM sodium ascorbate, store frozen at –20°C.
4. Stock solution of 1 mM Fe(II) 2 mM EDTA; store frozen at –20°C.
Cleavage of DNA by Histone–Fe(II) EDTA 279
5. Solution of 0.15% H
2
O
2
, freshly made.
6. Stop solution: 50% glycerol and 10 mM EDTA.
7. Microcentrifuge filtration devices (Series 8000 can be obtained from Lida Manu-
facturing Corporation).
8. 10 mM Tris-HCl, pH 8.0, and 0.1% SDS.
9. Microcentrifuge pestles, can be obtained from Stratagene.
10. 95% and 70% ethanol solutions, cooled to –20°C.
11. 3 M sodium acetate.
2.7. Sequencing Gel Analysis
1. Solid urea; molecular biology grade.
2. 5X TBE.
3. 40% Acrylamide (19:1 acrylamide:bis-acrylamide).
4. 20% APS (ammonium persulfate).
5. TEMED (N,N,N',N'-tetramethylethylenediamine).
6. 1 mL formamide loading buffer: 100% formamide, 0.05% bromophenol blue,
0.05% xylene cyanol, and 1 mM EDTA.
3. Methods
3.1. Overexpression and Purification of Single-Cysteine-
Substituted Proteins
The following methods work well for incorporating a single amino acid sub-
stitution into any protein of interest. Standard three or four primer PCR meth-
ods are used.
1. Standard PCR methods are used to amplify a DNA fragment containing a cysteine
codon in place of the wild-type codon. If the codon to be changed is near the end
of the amplified coding region, then only two primers are necessary with the
change incorporated into one of these “parent” primers. If more central to the
sequence, then a 3 primer technique is used with the change incorporated into an
internal primer, amplify with the internal primer and one of the parent primers
and then use the short amplified fragment as a primer with the remaining parent
primer and the original DNA as the template. Finally, if this method fails, two
complementary internal primers with the intended change are used to amplify
overlapping short fragments using the appropriate parent primers; these two frag-
ments are then combined with the parent primers and the entire insert amplified
without an additional template added.
2. Gel purify the DNA insert of interest. We typically use the electroelution tech-
nique after separating the PCR DNA on a 1% agarose gel. A slice of agarose
containing the insert of interest is placed into a dialysis membrane with enough
TBE to cover the agarose. Both ends of the dialysis membrane are clipped shut
and placed into a standard DNA electrophoresis apparatus containing 1X TBE
buffer; the DNA electroeluted is for 15 min at 150 V. Remove the TBE buffer
from the dialysis membrane containing the PCR DNA and precipitate.
280 Chafin and Hayes
3. Ligate the insert containing the single cysteine substitution into the appropriate
expression vector. We typically use the pET expression system (Novagen). Both
DNAs must be digested with the same restriction endonucleases. Incubate
equimolar amounts of insert DNA and pET3d DNA in 1X T
4
ligation buffer.
Add 400 U of T
4
DNA ligase (Bio-Labs) and incubate at 4°C overnight (see Note 2).
4. Check the efficiency of the ligation by transforming a small amount of the liga-
tion sample into DH5a cells. Place the transformation on LB–ampicillin plates
and incubate at 37°C overnight.
5. Prepare DNA from several colonies by placing a single colony into 3–5 mL of
LB–ampicillin medium and grow at 37°C. Isolate the DNA from these cultures
by standard DNA mini-prep techniques (see Note 3).
6. Digest part of the isolated plasmid with the original restriction endonucleases
used for ligation to liberate the DNA fragment corresponding to the original
insert. The plasmids that contain correct inserts can be used to transform BL21
cells for overexpression.
7. Transform the pET plasmid containing the insert into BL21 cells in the same
manner as for the DH5a cells (see step 4).
8. Place one BL21 colony from the LB–ampicillin plate into 200 mL of LB–ampi-
cillin medium.
9. Grow the culture in the absence of IPTG at 37°C to an optical density of 0.6 at
595-nm wavelength light. Add IPTG to a final concentration of 0.2 mg/mL and
return the culture to 37°C for approx 2–4 h (see Note 4).
10. After 2–4 h, pellet the bacteria by centrifugation at 4000g for 15 min.
11. Decant the supernatant and resuspend the pellet in 5–10 mL of TE buffer.
12. Add 10 mg/mL lysozyme to a final concentration of 0.2 mg/mL. Then, add
Triton-X 100 to a final concentration of 0.2% and incubate for 30 min at
room temperature.
13. Dilute the bacteria twofold with 2 M NaCl to a final concentration of 1 M NaCl.
Transfer the bacteria to Oakridge centrifuge tubes on ice.
14. Sonicate the bacterial slurry for 6 min total in two 3-min sonication steps (see
Note 5).
15. Pellet the cell debris by centrifugation at 10,000g for 30 min at 4°C.
16. Add PMSF to a final concentration of 0.1 mM. Dilute the supernatants twofold
with TE buffer to bring the NaCl concentration to 0.5 M.
17. Incubate the diluted supernatant with 12.5 mL of a 50% suspension of Bio-Rex
70-mesh beads for 4 h at 4°C with rotation. Linker histones and most other pro-
teins will bind directly to the Bio-Rex 70 beads. However, core histone proteins
must first be incubated with their dimerization partner proteins before they will
bind to the chromatography matrix (i.e., H2A with H2B) (see Note 6).
18. After 4 h collect the beads in a plastic 10-mL disposable chromatography col-
umn. Collect the flowthrough fraction in a 50-mL conical tube and freeze.
19. Wash the column with 2–3 column volumes of 10 mM Tris-HCl, pH 8.0 contain-
ing 0.6 M NaCl. Collect the first 10 mL of the wash fraction in a 15-mL conical
tube and freeze.
Cleavage of DNA by Histone–Fe(II) EDTA 281
20. Elute the bound proteins with two separate 1-column volume elution steps of 10 mM
Tris-HCl, pH 8.0 containing 1.0 M NaCl. Collect the 1.0 M elution steps in sepa-
rate 15 mL conical tubes and freeze.
21. After elution, wash the column with one column volume of 10 mM Tris-HCl,
pH 8.0 containing 2.0 M NaCl. Collect the 2.0 M elution step in a 15-mL conical
tube and freeze.
22. Check 10 µL of each fraction for protein by loading a small amount on a 12% or
18% SDS–polyacrylamide gel.
3.2. Reduction and Modification of Cysteine-Substituted Proteins
3.2.1. Reduction of Cysteine-Substituted Proteins
1. Incubate protein of interest in a 15-mL conical tube with 50 mM DTT final con-
centration for 1 h on ice.
2. Dilute the protein sample twofold with TE; this dilutes the NaCl concentration to
500 mM NaCl.
3. Add 0.8 mL of a 50% slurry of Bio-Rex 70 chromatography resin and incubate
for 2 h at 4°C with rotation. We have found that Bio-Rex 70 can bind approx 0.5 mg
protein/g resin.
4. Pour slurry into a 10-mL plastic, disposable chromatography column and collect
the flowthrough fraction. Disposable chromatography columns are commercially
available from Bio-Rad or other manufacturers.
5. Wash the column with 3–5 column volumes of buffer containing 10 mM Tris-HCl,
pH 8.0, and 0.5 M NaCl. Immediately remove 20 µL of the freshly eluted wash
sample into a separate Eppendorf tube for analysis later on a 12% SDS–polyacryla-
mide gel and immediately freeze the larger sample in case the protein did not bind
the resin. Aliquoting the sample in this manner ensures that the sample does not
need to be thawed for analysis.
6. An intermediate wash of the column with buffer containing 0.6 M NaCl is per-
formed to remove proteins that are less well-bound because of partial degrada-
tion. Aliquots of these samples are obtained in the same manner as the previous
wash step.
7. Linker histone protein can be eluted with 1-column volume steps of the same
buffer except with 1.0 M NaCl. Typically, five separate 1-column volume 1.0 M
NaCl elution steps are performed and collected separately. Usually, only 5 µL of
the elution steps needs be aliquoted for SDS–polyacrylamide gel analysis. As
above, the large elution fractions are frozen immediately. A final elution with
buffer containing 2.0 M NaCl buffer will ensure that all of the protein has been
eluted from the column.
8. Check the protein content of each aliquot obtained from the wash and elution
steps on a 12% SDS–polyacrylamide gel. After separation, incubate the protein
gel in enough Coomassie blue stain to cover the protein gel. Stain for approx 1 h
at room temperature and destain with 45% methanol and 10% acetic acid until
the background of the gel is clear.
282 Chafin and Hayes
3.2.2. Modification of Cysteine Substituted Proteins with EPD
1. Thaw the fraction containing the reduced protein to be modified with EPD on ice.
Working as quickly as possible, add a 1.1 fold molar excess of EPD to 60 µL
(approx 30 µg) of reduced protein. Incubate for 1 h at room temperature in
the dark.
2. Removal of excess cleavage reagent requires one more round of Bio-Rex 70 chro-
matography, identical to that in step 3 of Subheading 3.2.1. except that 60 µL of
the 50% slurry is added to the protein. In addition, the slurry is poured into a
column made from a blue 1-mL pipet tip fitted with glass wool at the opening.
Wash and elute as in step 5 of Subheading 3.2.1. except all elution volumes are
scaled according to the resin amount. Aliquots for protein analysis are exactly the
same size as previously indicated.
3. Postmodification labeling with
14
C-NEM (N-[ethyl–1–
14
C]-maleimide) (New
England Nuclear) can be used to quantitatively determine the extent of modifica-
tion with the DNA cleavage reagent. Add 0.25–0.5 µCi of
14
C-NEM to each
protein aliquot made from the elution fractions of the Bio-Rex column. After
10 min, add 2 volumes of 2X protein loading buffer and separate the proteins
on a 12% SDS-polyacrylamide gel. Stain and destain the gel as in step 8 of
Subheading 3.2.1. and dry the gel onto a piece of Whatman filter paper. Visual-
ize the labeled proteins by exposing the dried gel to ultra sensitive Bio-Max auto-
radiography film.
4. A protein gel at this step performs two functions. (1) It determines which frac-
tions contain the protein of interest and (2) it determine the extent of modifica-
tion with the DNA cleavage reagent.
3.3. Radioactive End-Labeling
of a Purified DNA Restriction Fragment
1. Treat approx 5 µg of plasmid DNA or 1 µg of a purified DNA fragment with the
appropriate restriction endonuclease in the manufacturer’s buffer.
2. Precipitate the DNA by adjusting the solution to 0.3 M sodium acetate and add-
ing of 2.5 vol of cold ethanol.
3. Resuspend the DNA in phosphatase buffer and treat with alkaline phosphatase
for 1 h at 37°C.
4. Adjust the solution to 0.1% SDS, phenol extract the solution, and then precipitate
the aqueous phase twice with ethanol and sodium acetate.
5. Resuspend the DNA in 10 µL TE and add 2.5 µL of 10X T
4
polynucleotide
kinase buffer.
6. Add 50 µCi of [γ-
32
P]dATP and adjust the volume to 24 µL with water.
7. Start the reaction by adding 10 units of T
4
polynucleotide kinase and incubate for
30 min at 37°C.
8. Stop the kinase with 200 µL of 2.5 M ammonium acetate (NH
4
Oac) and 700 µL
of cold 95% ethanol.
9. Pellet the DNA in a microcentrifuge for 30 min at room temperature.
Cleavage of DNA by Histone–Fe(II) EDTA 283
10. Wash the DNA pellet briefly with cold 70% ethanol and dry the DNA in a
Speedvac concentrator.
11. Dissolve the DNA in 34 µL of TE buffer.
12. Digest the DNA fragment with a second restriction endonuclease that liberates
the fragment of interest and yields fragments that can be easily separated on a
native 6% polyacrylamide gel.
13. After separation, wrap the gel tightly in plastic wrap and apply fluorescent mark-
ers onto various portions of the gel for alignment purposes (can be obtained from
Stratagene) or accurately mark the position of the gel on the film. Expose the wet
gel to the autoradiography film for 1 min, which is sufficient to detect the spe-
cific band containing the labeled fragment.
14. Excise the band of interest from the polyacrylamide gel and place into a clean
Eppendorf tube. Crush the acrylamide gel slice with a Eppendorf pestle and
add 700 µL of TE buffer. The labeled fragment will elute overnight with pas-
sive diffusion.
15. Split the sample equally into two Series 8000 Microcentrifuge Filtration Devices
and spin for 30 min in a microcentrifuge.
16. Precipitate the eluted DNA and dissolve in TE buffer pH 8.0. Add enough TE
buffer so that the labeled DNA is approx 1000 cpm/µL (see Note 7).
3.4. Reconstitution of Nucleosomes by Salt Step Dialysis
The method described here for the reconstitution of nucleosomes allows for
large quantities of nearly homogeneous core particles in 12 h (13). Moreover,
reconstituted nucleosomes are known to bind linker histone in a physiologi-
cally relevant manner according to multiple criteria. Virtually any piece of
DNA 147 bp or longer can be used. However to obtain nucleosomes with only
one translational position, the DNA sequence should contain nucleosome posi-
tioning sequences such as that from the Xenopus borealis somatic 5S rRNA
gene (14–16). The DNA can be labeled on the 5' or 3' end with commercially
available enzymes after phosphatase treatment as described above.
1. Add approx 5–8 µg of unlabeled calf thymus DNA, 200,000–400,000 cpm of
singly labeled Xenopus borealis 5S ribosomal DNA, purified chicken erythro-
cyte core histone protein fractions (H2A/H2B and H3/H4) (see Note 1), 160 µL
of 5 M NaCl (2.0 M final), and TE buffer to a final of volume 400 µL.
2. Place the reconstitution mixture into a 6 to 8 kDa molecular weight cut-off dialy-
sis bag. All subsequent dialysis steps are for 2 h at 4°C against 1 L of dialysis
buffers unless specified. The first dialysis buffer is 10 mM Tris-HCl, pH 8.0, 1.2 M
NaCl and 1 mM EDTA. Subsequent dialyses steps are with fresh buffer contain-
ing 1.0 M, 0.8 M, and then 0.6 M NaCl. The procedure is completed with a final
dialysis against TE buffer overnight. Nucleosomes at this stage can be used for
gel-shift experiments where EDTA does not interfere.
3. For DNA cleavage experiments with EPD, two additional dialysis steps are
required. First dialyze the reconstitutes against 10 mM Tris-HCl, pH 8.0 several
284 Chafin and Hayes
hours to remove the EDTA. A second dialysis against fresh 10 mM Tris-HCl,
pH 8.0 removes trace amounts of EDTA and prepares the samples for chemical
mapping with EPD.
3.5. Maxim–Gilbert G-Specific Reaction
The G-specific reaction used in the Maxim–Gilbert sequencing method pro-
vides an easy and quick method to identify the exact location of bases within
any known sequence on sequencing gels. It is used here to determine the sites
of DNA to base-pair resolution. Because this method is not generally used any
longer, the steps are outlined as follows:
1. Add approx 20,000 cpm of singly labeled DNA (same DNA used to reconsti-
tute nucleosomes).
2. Add 20 µL of 10X G-specific reaction buffer.
3. Add water to a final volume of 200 µL.
4. Start by adding 1 µL of straight dimethylsulfate (DMS) to the tube. Mix immedi-
ately and spin briefly in a microfuge (do this in a hood; be careful not to get any
DMS on your skin or on standard laboratory gloves. Store DMS in a tightly
capped brown glass bottle at 4°C).
5. Add 50 µL of G-reaction stop solution and mix immediately.
6. Add 2.5 vol of –20°C 95% ethanol to precipitate the DNA.
7. Wash the DNA with –20°C ethanol; dry and dissolve the DNA in 90 µL of H
2
O.
8. Add 10 µL of piperidine and incubate at 90°C for 30 min.
9. Dry the DNA solution in a Speedvac to completion.
10. Dissolve the DNA in 20 µL of water and repeat the drying step. Repeat this step
one more time.
11. Dissolve DNA in 100 µL TE buffer and store at 4°C.
3.6. Site-Directed Hydroxyl Radical Cleavage of DNA
3.6.1. Binding Single-Cysteine-Substituted Linker Histone Proteins
to Reconstituted Nucleosomes
1. The exact amount of each mutant linker histone protein needed to stoichiometri-
cally bind the nucleosome needs to be determined empirically. Increasing
amounts of the linker histone are titrated to a fixed volume of reconstituted
nucleosomes (typically 5000 cpm) and analyzed via a gel-shift procedure (17).
This is typically scaled up 10-fold for the site-specific cleavage reaction.
2. Add 5% glycerol final to the binding reaction (analytical scale only, site-specific
cleavage reactions contain 10-fold less glycerol).
3. Add 50 mM NaCl final to the binding reaction (see Note 8).
4. Incubate the binding reactions for 15 min at room temperature.
5. Separate the complexes on a 0.7% agarose and 0.5X TBE gel. After drying the
gel, expose to autoradiograpic film and determine the amount of protein neces-
sary for good complex formation.