King M.R., Mody N.A. Numerical and Statistical Methods for Bioengineering: Applications in MATLAB
Подождите немного. Документ загружается.


match perfectly. It is similar in magnitude to PAM250, but has been determined to be
more reliable for protein sequences.
When applying a scoring matrix to determine an optimal alignment between two
sequences, a brute-force search of all possible alignments would in most cases be
prohibitive.
For instance, suppose one has two sequences of length n. If we add some number
of gaps such that (i) each of the two sequences has the same length and (ii) a gap
cannot align with another gap, it can be shown that, as n → ∞, the number of possible
alignments approaches
C
2n
n
2
2n
ffiffiffiffiffi
nπ
p
which for n = 100 takes on the astronomical sum of 10
59
!
To address this, Needleman and Wunsch (1970) first app lied the dynamic
programming concept from computer science to the pr oblem of aligning two
protein sequences. They defined a maximum match as the largest number of
amino acids of one protein (X) that can be matched with those of a nother protein
(Y) while allowing for all possible deletions. The maximum match is found with a
matrix in which all possible pair combinations that can be formed from the amino
acid sequences are repres ented. The rows and columns of this matrix are associated
with the ith and jth amino acids of proteins X and Y, respectively. All of the
possible comparisons are then represented by pathways through the two-
dimensional matrix. Every i or j can only occur once in a pathway because a
particular amino acid cannot occupy multiple positions at once. A pathway can be
denoted as a line connecting elements of the matrix, with complete diagonal
pathways comprising no gaps.
As first proposed by Needleman and Wunsch, each viable pathway through the
matrix begins at an element in the first column or row. Either i or j is increased by
exactly one, while the other index is increased by one or more; in other words, only
diagonal (not horizonta l or vertical) moves are permitted through the matrix. In this
manner, the matrix elements are connected in a pathway that leads to the final
element in which either i or j (or both simultaneously) reach their final values. One
can define various schemes for rewarding amino acid matc hes or penalizing mis-
matches. Perhaps the simplest method is to assign a value of 1 for each match and a
value of 0 for each mismatch. More sophisticated scoring methods can include the
influence of neighboring residues, and a penalty factor for each gap allowed. This
approach to sequence alignment can be generalized to the simultaneous comparison
of several proteins, by extending the two-dimensional matr ix to a multidimensional
array.
The maximum-match operation is depicted in Figure 9.2, where 12 amino acids
within the hydrophobic domain of the protein β-synuclein (along the vertical axis, i)
are compared against 13 amino acids of the α-synuclein sequence (hori zontal axis, j).
Note that α-synuclein (SNCA) is primarily found in neural tissue, and is a major
component of pathological regions associated with Parkinson’s and Alzheimer’s
diseases (Giasson et al., 2 001). An easy way to complete the depicted matrix is to
start in the last row, at the last column position. The number within ea ch element
represents the maximum total score when starting the alignment with these two
amino acids and proceeding in the positive i, j directions. Since, in this final position
A = A, we assign a value of 1. Now, staying in the final row and moving towards the
left (decreasing the j index), we see that starting the final amino acid of β-syn at any
547
9.2 Sequence alignment and database searches

other position of α-syn results in no possible matches, and so the rest of this row fills
out as zeros. Moving up one row to the i = 11 posit ion of β-syn, and proceeding
from right to left, we see that the first partial alignment that results in matches is EA
aligned with EA, earning a score of 2. Moving from right to left in the decreasing j
direction, we see the maximum score involving only the last two amino acids of β-syn
vary between 1 and 2. Moving up the rows, we may continue complet ing the matrix
by assigning a score in each element corresponding to the maximum score obtained
when starting at that position and proceeding in the positive i and j directions while
allowing for gaps. The remainder of the matrix can be calculated in a similar fashion.
The overall maximum matc h is then initiated at the greatest score found in the first
row or column, and a pathway to form this maximum match is traced out as depicted
in Figure 9.2. As stated previously, no horizont al or vertical moves are permitted.
Although not depicted here, it is sometimes possible to find alternate pathways with
the same overall score. In this example, the maximum match alignment is de ter-
mined to be as follows:
α-syn: PSEEGYCDYEPEA
β-syn: PQEE–YCEYEPEA
*&*
where the asterisks indicate mismatches, and a gap is marked with an ampersand.
A powerful and popular algorithm for sequence comparison based on dynamic
programming is called the Basic Local Alignment Search Tool,orBLAST. The first
step in a BLAST search is call ed “seeding,” in which for each word of length W in the
query, a list of all possible words scoring within a threshold T are generated. Next, in
an “extension” step, dynamic programming is used to extend the word hits until the
score drops by a value of X. Finally, an “evaluation” step is performed to weigh the
significance of the extended hits, an d only those above a predefined threshold are
Figure 9.2.
Comparison of two sequences using the Needleman–Wunsch method.
0000000000001
1122111112120
3222222222310
3344333334220
4444454453210
5566555544220
6665556543210
7776676553210
7788765544210
8898765544220
9987765543210
10
A
E
P
E
Y
E
C
Y
E
E
Q
P 987765543310
P SEEGYCDYEPEA
548
Basic algorithms of bioinformatics

reported. A variation on the BLAST algorithm, which includes the possibility of two
aligned sequences connected with a gap, is called BLA ST2 or Gapped BLA ST. It
finds two non-overlapping hits with a score of at least T and a maximum distance d
from one another. An ungapped extension is performed and if the generated highest-
scoring segmen t pairs (HSP) have sufficiently high scores when normalized appro-
priately by the segment length, then a gapped extension is initiated. Results are
reported for alignments showing sufficiently low E-value. The E-value is a statistic
which gives a measure of whether pot ential matches might have arisen by chance. An
E-value of 10 signifies that ten matches would be expected to occur by chance. The
value 10 as a cutoff is often used as a default, and E < 0.05 is generally taken as being
significant. Generally speaking, long regions of moderate similarity are more sig-
nificant than short regions of high identity. Some of the specific BLA ST programs
available at the NIH Natio nal Center for Biotechnology Inform ation (NCBI, http://
blast.ncbi.nlm.nih .gov/Blast.cgi) include: blastn, for submitting nucleotide queries
to a nucleotide database; blastp, for protein queries to the protein database; blastx,
for searching protein databases using a translated nucleotide query; tblastn, for
searching translated nucleotide databases using a protein query; and tblastx, for
searching translated nucleotide databases using a translated nucleotide que ry.
Further variations on the original BLAST algorithm are available at the NCBI
Blast query page, and can be selected as options. PSI-BLAST, or Position-Specific
Iterated BLAST, allows the user to build a position-specific scoring matrix using the
results of an initial (default option) blastp query. PHI-BLAST, or Pattern Hit
Initiated BLAST, finds pro teins which contain the pattern and similarity within
the region of the pattern, and is integrated with PSI-BLAST (Altschul et al., 1997).
There are many situations where it is desirable to perform sequence comparison
between three or more proteins or nucleic acids. Such comparisons can help to identify
functionally important sites, predict protein structure, or even start to reconstruct the
evolutionary history of the gene. A number of algorithms for achieving multiple
sequence alignment are available, falling into the categories of dynamic programming,
progressive alignment,oriterative search methods. Dynamic programming methods
proceed as sketched out above. The classic Needleman–Wunsch method is followed
with the difference that a higher-dimensional array is used in place of the two-
dimensional matrix. The number of comparisons increases exponentially with the
number of sequences; in practice this is dealt with by constraining the number of letters
or words that must be explicitly examined (Carillo and Lipman, 1988). In the pro-
gressive alignment method, we start by obtaining an optimal pairwise alignment
between the two most similar sequences among the query group. New, less related
sequences are then added one at a time to the first pairwise alignment. There is no
guarantee following this method that the optimal alignment will be found, and the
process tends to be very sensitive to the initial alignments. The ClustalW (“Weighted”
Clustal; freely available at www.ebi.ac.uk/clustalw or align.genome.jp among other
sites) algorithm is a general purpose multiple sequence alignment program for DNA
or proteins. It performs pairwise alignments of all submitted sequences (maximum
sequence no. = 30; maximum length = 10 000) and then produces a phylogenetic
tree (see Section 9.3 ) for comparing evolutionary relationships (Thompson et al.,
1994). Sequences are submitted in FASTA format, and the newer versions of
ClustalW can produce graphical output of the results. T-Coffee (www.ebi.ac.uk/
t-coffee or www.tcoffee.org) is another multiple sequence alignment program, a
progressive method which combines information from both local and global align-
ments (Notredame et al., 2000 ). This helps to minimize the sensitivity to the first
549
9.2 Sequence alignment and database searches

alignments. Another benefit of the T-Coffee program is that it can combine partial
results obtained from several different alignment methods, such as one alignment
from ClustalW, another from a program called Dialign, etc., and will combine this
information to produce a new multiple sequence alignment that integrates this
Box 9.3 BLAST search of a short protein sequence
L-selectin is an adhesion receptor which is important in the trafficking of leukocytes in the bloodstream.
L-selectin has a short cytoplasmic tail, which is believed to perform intracellular signaling functions and
help tether the molecule to the cytoskeleton (Green et al., 2004; Ivetic et al., 2004). Let’s perform a
simple BLAST search on the 17 amino acid sequence encoding the human L-selectin cytoplasmic tail,
RRLKKGKKSKRSMNDPY, to see if any unexpected connections to other similar protein sequences can
be revealed. Over the past ten years, performing such searches has become increasingly user friendly
and simple. From the NCBI BLAST website, we choose the “protein blast” link. Within the query box,
which requests an accession number or FASTA sequence, we simply paste the 17-AA code and then
click the BLAST button to submit our query. Some of the default settings which can be viewed are: blastp
algorithm, word size = 3, Expect threshold = 10, BLOSUM62 matrix, and gap costs of −11 for gap
creation and −1 for gap extension. Within seconds we are taken to a query results page showing 130
Blast hits along with the message that our search parameters were adjusted to search for a short input
sequence. This automatic parameter adjustment can be disabled by unchecking a box on the query
page. All non-redundant GenBank CDS translations + PDB + SwissProt + PIR + PRF excluding
environmental samples from WGS projects have been queried.
3
Table 9.4 presents the first 38 hits,
possessing an E-value less than 20.
The first group of letters and numbers is the Genbank ID and accession number (a unique number
identified for sequence entries), and in the Blast application these are hyperlinks that take you to the
database entry with sequence data, source references, and annotation into the protein’s known
functions. The first 26 hits are all for the L-selectin protein (LECAM-1 was an early proposed name),
with the species given in square brackets. The “PREDICTED” label indicates a genomic sequence that is
believed to encode a protein based on comparison with known data. Based on the low E-value scores of
the first 20+ hits, the sequence of the L-selectin cytoplasmic tail is found to be highly conserved among
species, perhaps suggesting its evolutionary importance. Later we will explore a species-to-species
comparison of the full L-selectin protein. The first non-L-selectin hit is for the amphioxus dachshund
protein in a small worm-like creature called the Florida lancelet. Clicking on the amphioxus dachshund
score of 33.3 takes us to more specific information on this alignment:
>gb|AAQ11368.1| amphioxus dachshund [Branchiostoma floridae]
Length = 360
Score = 33.3 bits (71), Expect = 2.8
Identities = 10/12 (83%), Positives = 11/12 (91%), Gaps = 0/12 (0%)
Query 2 RLKKGKKSKRSM 13
RLKKGKK+KR M
Sbjct 283 RLKKGKKAKRKM 294
Note that the matched sequence only involves 12 of the 17 amino acids from the L-selectin
cytoplasmic tail, from positions 2 to 13. In the middle row of the alignment display, only matching
letters are shown and mismatches are left blank. Conservative substitutions are shown by a + symbol.
Although no gaps were inserted into this particular alignment, in general these would be indicated by a
dash. The nuclear factor dachshund (dac) is a key regulator of eye and leg development in Drosophila. In
man, it is a retinal determination protein. However, based on the relatively large E-score and short length
for this and other non-L-selectin hits, in the absence of any other compelling information it would be
unwise to overinterpret this particular match.
3
These terms refer to the names of individual databases containing protein sequence data.
550
Basic algorithms of bioinformatics

Table 9.4. Output data from BLAST search on L-selectin subsequence
Score E-value
dbj|BAG60862.1| unnamed protein product [Homo sapiens] 57.9 1e−07
emb|CAB55488.1| L-selectin [Homo sapiens] >emb|CAI19356.1| se ... 57.9 1e −07
gb|AAC63053.1| lymph node homing receptor precursor [Homo sap ... 57.9 1e−07
emb|CAA34203.1| pln homing receptor [Homo sapiens] 57.9 1e−07
emb|CAB43536.1| unnamed protein product [Homo sapiens] >prf|| ... 57.9 1e−07
ref|NP_000646.1| selectin L precursor [Homo sapiens] >sp|P141 ... 57.9 1e−07
emb|CAA34275.1| unnamed protein product [Homo sapiens] 57.9 1e−07
ref|NP_001009074.1| selectin L [Pan troglodytes] >sp|Q95237.1 ... 55.4 6e−07
ref|NP_001106096.1| selectin L [Papio anubis] >sp|Q28768.1|LY ... 52.0 7e−06
ref|NP_001036228.1| selectin L [Macaca mulatta] >sp|Q95198.1| ... 52.0 7e−06
sp|Q95235.1|LYAM1_PONPY RecName: Full=L-selectin; AltName: Fu ... 52.0 7e−06
ref|NP_001106148.1| selectin L [Sus scrofa] >dbj|BAF91498.1| ... 47.7 1e−04
ref|XP_537201.2| PREDICTED: similar to L-selectin precursor ( ... 44.8 0.001
ref|XP_862397.1| PREDICTED: similar to selectin, lymphocyte i ... 44.8 0.001
ref|XP_001514463.1| PREDICTED: similar to L-selectin [Ornitho ... 43.9 0.002
gb|EDM09364.1| selectin, lymphocyte, isoform CRA_b [Rattus no ... 40.1 0.026
ref|NP_001082779.1| selectin L [Felis catus] >dbj|BAF46391.1| ... 40.1 0.026
ref|NP_062050.3| selectin, lymphocyte [Rattus norvegicus] >gb ... 40.1 0.026
dbj|BAE42834.1| unnamed protein product [Mus musculus] 40.1 0.026
dbj|BAE42004.1| unnamed protein product [Mus musculus] 40.1 0.026
dbj|BAE37180.1| unnamed protein product [Mus musculus] 40.1 0.026
dbj|BAE36559.1| unnamed protein product [Mus musculus] 40.1 0.026
sp|P30836.1|LYAM1_RAT RecName: Full=L-selectin; AltName: Full ... 40.1 0.026
ref|NP_035476.1| selectin, lymphocyte [Mus musculus] >sp|P183 ... 40.1 0.026
gb|AAN87893.1| LECAM-1 [Sigmodon hispidus] 39.7 0.034
ref|NP_001075821.1| selectin L [Oryctolagus cuniculus] >gb|AA ... 34.6 1.2
gb|AAQ11368.1| amphioxus dachshund [Branchiostoma floridae] 33.3 2.8
ref|XP_001491605.2| PREDICTED: similar to L-selectin [Equus c ... 32.0 6.8
ref|YP_001379300.1| alpha-L-glutamate ligase [Anaeromyxobacte ... 31.2 12
ref|NP_499490.1| hypothetical protein Y66D12A.12 [Caenorhabdi ... 31.2 12
ref|XP_002132434.1| GA25185 [Drosophila pseudoobscura pseudoo ... 30.8 16
ref|YP_001088484.1|
ABC
transporter, ATP-binding protein [Clo ... 30.8 16
ref|ZP_01802998.1| hypothetical protein CdifQ_04002281 [Clost ... 30.8 16
gb|AAF34661.1|AF221715_1 split ends long isoform [Drosophila ... 30.8 16
gb|AAF13218.1|AF188205_1 Spen RNP motif protein long isoform ... 30.8 16
ref|NP_722616.1| split ends, isoform C [Drosophila melanogast ... 30.8 16
ref|NP_524718.2| split ends, isoform B [Drosophila melanogast ... 30.8 16
ref|NP_722615.1| split ends, isoform A [Drosophila melanogast ... 30.8 16
551
9.2 Sequence alignment and database searches

information. T-Coffee tends to obtain better results than ClustalW for sequences
with less than 30% identity, but is slower. Figure 9.3 shows a multiple sequence
alignment using ClustalW of eight species (human, chimpanzee, orangutan, rhesus
monkey, domestic dog, house mouse, Norway/brown rat, and hispid cotton rat) of
Figure 9.3.
Multiple sequence alignment of L-selectin protein sequence from eight different species, generated using the ClustalW
program. Columns with consensus residues are shaded in black, with residues similar to consensus shaded in gray. The
consensus sequence shows which residues are most abundant in the alignment at each position.
552
Basic algorithms of bioinformatics

Figure 9.4.
Multiple sequence alignment of L-selectin protein sequence from seven different species, generated using the TCoffee program. Consensus regions are shaded
in grey and marked with asterisks.

L-selectin protei n, processed using BoxShade (a commonly used simple plotting
program). ClustalW and BoxShade programs were accessed at the Institute Pasteur
website. Figure 9.4 shows a multiple sequence alignment performed via the T-Coffee
algorithm at the Swiss EMBnet website.
Figures 9.3 and 9.4 demonstrate that the N-terminal binding domain, and the
C-terminal transmembrane and cytoplasmic domains, are the most highly conserved
between species. The intermediate epidermal growth factor-like domain, two short
consensus repeat sequences, and short spacer regions show more variability and are
perhaps not as important biologically.
A third category of iterative methods seek to increase the multiple sequence align-
ment score by making random alterations to the alignment. The first step is to obtain a
multiple sequence alignment, for instance from ClustalW. Again, there is no guarantee
that an optimal alignment will be found. Some examples of iterative methods include:
simulated annealing (MSASA; Kim et al., 1994); genetic algorithm (SAGA; Notredame
and Higgins, 1996); and hidden Markov model (SAM; Hughey and Krogh, 1996).
9.3 Phylogenetic trees using distance-based methods
Phylogenetic trees can be used to reconstruct genealogies from molecular data, not
just for genes, but also for species of organisms. A phylogenetic tree, or dendrogram,
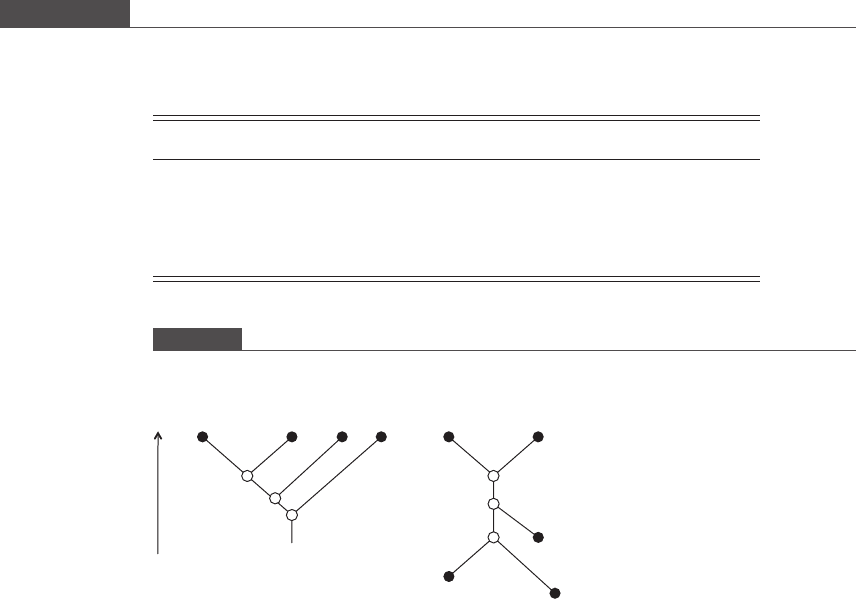
is an acyclic two-dimensional graph of the evolutionary relationship among three or
more genes or organisms. A phylogenetic tree is composed of nodes and branches
connecting them. Terminal nodes (filled circles in Figure 9.5), located at the ends of
branches, represent genes or organisms for which data are available, whereas inter-
nal nodes (ope n circles) represent inferred common ancestors that have given rise to
two independent lineages. In a scaled tree, the lengths of the branches are made
proportional to the differences between pairs of neighboring nodes, whereas in
unscaled trees the branch lengths have no significance. Some scaled trees are also
additive, meaning that the relative number of accumulated differences between any
two nodes is represented by the sum of the branch lengt hs. Figure 9.5 also shows the
distinction between a rooted tree and an unrooted tree. Rooted trees possess a
common ancestor (iii) with a unique path to any “leaf”, and tim e progresses in a
clearly defined direction, whereas unrooted trees are undirected with a fewer number
of possible combinations. The number of possible trees (N) increases sharply with
increasing numb er of species (n), and is given by the following expressions for rooted
and unrooted trees, respectively:
N
R
¼
2n 3ðÞ!
2
n2
n 2ðÞ!
;
N
U
¼
2n 5ðÞ!
2
n3
n 3ðÞ!
:
Table 9.5 shows the number of possible phylogenetic trees arising from just 2–16
data sets.
Clustering algorithms are algorithms that use the distance (number of mutation
events) to calculate phylogenetic trees, with the trees based on the relative numbers of
similarities and differences between sequences. Distance matrices can be constructed by
computing pairwise distances for all sequences. Sequential clustering is a technique in
which the data sets are then linked to successively more distant taxa (groups of related
554
Basic algorithms of bioinformatics
organisms). Distances between pairs of DNA sequences are relatively simple to com-
pute, and are given by the sum of all base pair differences between the two sequences.
The pairs of sequences to be compared must be similar enough to be aligned. All base
changes are treated equally, and insertions/deletions are usually weighted more heavily
than replacements of a single base, via a gap penalty. Distance values are often
normalized as the “number of changes per 100 nucleotides.” In comparison, amino
acid distances are more challenging to compute. Different substitutions have varying
effects on structure and function, and some amino acid substitutions of course require
greater than one DNA mutation (see Table 9.1). Replacement frequencies are provided
by the PAM and BLOSUM matrices, as discussed previously.
The originally proposed distance matrix method, which is simple to implement, is
called the unweighted-pair-group method with arithmetric mean (UPGMA; Sneath &
Sokal, 1973). UPGMA and other distance-based methods use a measure of all pair-
wise genetic distances between the taxa being considered. We begin by clustering the
two species with the smallest distance separating them into a new composite group.
For instance, if comparing species A through E, suppose that the closest pair is DE.
After the first cluster is formed, then a new distance matrix is formed between the
remaining species and the cluster DE. As an example, the distance between A and
the cluster DE would be calculated as the arithmetric mean d
A(DE)
=(d
AD
+ d
AE
)/2.
The species closest together in the new distance matrix are then clustered together to
form a new composite species. This process is continued until all species to be
compared are grouped. Groupings are then graphically represented by the phyloge-
netic tree, and in some cases the branch length is used to show the relative distance
between groupings. Box 9.4 gives an illustration of this sequential process.
Table 9.5. Number of possible rooted and unrooted phylogenetic trees
as a function of the number of data sets
No. of data sets, n No. of rooted trees, N
R
No. of unrooted trees, N
U
211
4153
8 135 135 10 395
16 6.19 × 10
15
2.13 × 10
14
Figure 9.5.
Examples of a rooted and unrooted phylogenetic tree. Terminal nodes are represented as filled circles and (inferred)
internal nodes are represented as open circles.
Rooted tree
(i)
A
A
B
C
D
E
(iii)
Unrooted tree
(ii)
(i)
BCD
(ii)
(iii)
Increasing time
555
9.3 Phylogenetic trees using distance-based methods

Box 9.4 Phylogenetic tree generation using the UPGMA method
Consider the alignment of five different DNA sequences of length 40 shown in Table 9.6. We first
generate a pairwise distance matrix to summarize the number of non-matching nucleotides between the
five sequences:
Species A B C D
B 13 –––
C 315––
D 8159–
E 714411
The unambiguous closest species pair is AC, with a distance of 3. Thus, our tree takes the following
form:
AC
We now generate a new pairwise distance matrix, treating the AC cluster as a single entity:
Species AC B D
B 14 ––
D 8.5 15 –
E 5.5 14 11
Thus, the next closest connection is between the AC cluster and E, with a distance of 5.5. Our tree
then becomes:
A
C
E
Table 9.6. Pairwise distance matrix from alignment of five sequences
10 20 30 40
A: TGCGCAAAAA CATTGCCCCC TCACAATAGA AATAAAGTGC
B: TGCGCAAACA CATTTCCTAA GCACGCTTGA ACGAAGGGGC
C: TGCGCAAAAC CATTGCCACC TCACAATAGA AATAAATTGC
D: TGCCCAAAAC CCTTGCGCCG TCACGATAGA AAAAAAGGGC
E: TGCGCAAAAC CATTTCCACG ACACAATCGA AATAAATTGC
556
Basic algorithms of bioinformatics