Heitjans P., Karger J. (Eds.). Diffusion in Condensed Matter: Methods, Materials, Models
Подождите немного. Документ загружается.

13 Viscoelasticity and Microscopic Motion in Dense Polymer Systems 539
a)
b)
Fig. 13.19. Dynamic structure factors from PE-melts at 509K (a) M
w
= 2000; (b)
M
w
= 12400. The solid lines display the predictions of the Rouse model.
are compared with the dynamic structure factor of the reptation model [47]
and are very well described. We note, that the fits were performed varying
only one single parameter, namely the tube diameter d, while the Rouse
rate was determined from earlier NSE data taken at short times. With this
one parameter it is possible to achieve quantitative agreement with respect
to both the Q and the time dependence of the dynamic structure factor.
We further note, that the dynamic structure factors predicted by the other
confinement models discussed above are not able to fit the data [16].
It is illustrative to visualize the large spread of the time regimes relevant
for the dynamics of the M
w
= 190000 molecule. The elementary Rouse time
amounts to 2.5 ps. The cross over time to local reptation τ
e
= 5 ns. The
Rouse time of the chain τ
R
=45µs and finally the disentanglement time
τ
d
=11.7 ms. Thus, the relevant time scales spread over a range of more than
nine orders of magnitude. The corresponding length scales are much closer
together. The segment length =
0
√
C
∞
,whereC
∞
is the characteristic
ratio, is 4
˚
A, the tube diameter d =46
˚
A and the chain end-to-end distance
R
E
= 418
˚
A.
We are now drawing a third conclusion. NSE experiments on long-chain
polymer melts have verified the molecular existence of an entanglement length
540 Dieter Richter
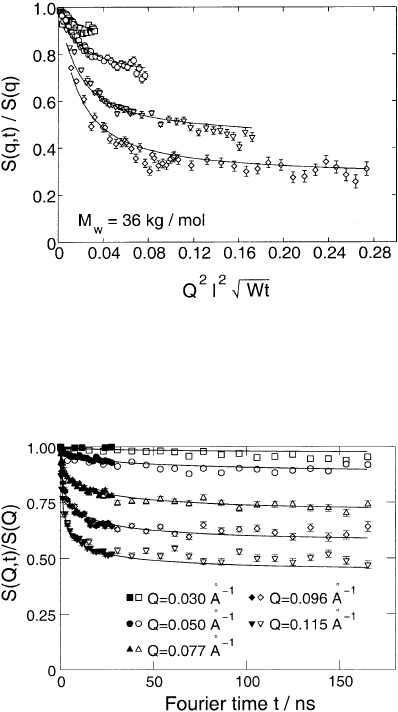
Fig. 13.20. Scaling presentation of the dynamic structure factor from a M
w
=
36000 PE-melt at 509 K as a function of the Rouse scaling variable. The solid lines
are a fit with the reptation model [47]. The Q-values (Q ≡ q) are (from above)
Q =0.05, 0.077, 0.115, 0.154
˚
A
−1
.
Fig. 13.21. Dynamic structure factor from a long-chain PE-melt (M
w
= 190000)
at 509 K. The solid lines represent a fit with the reptation model [47].
which, being of dynamic nature, under equilibrium only reveals itself in dy-
namic experiments. Moreover, the detailed Q-dependent dynamic structure
factor validates the idea of chain motion in a confining tube and corroborates
quantitatively the celebrated reptation model of de Gennes.
13.6 Intermediate Scale Dynamics
A precondition for the validity of the entropy-driven Rouse model is the re-
quirement that only chain connectivity enters into the equation of motion and
any influence from local stiffness or rotational barriers etc. are negligible. In
13 Viscoelasticity and Microscopic Motion in Dense Polymer Systems 541
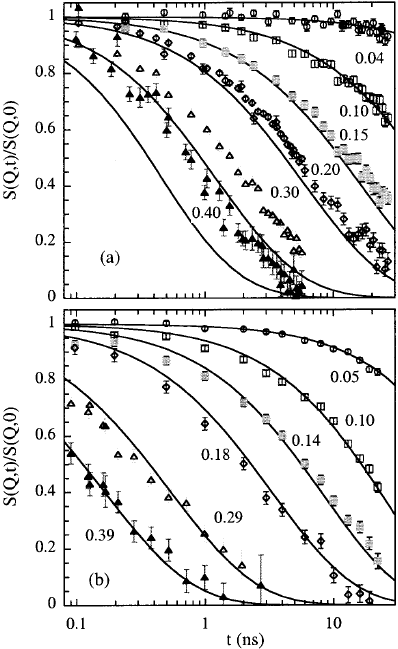
Fig. 13.22. Dynamic structure factor of (a)aPIBmelt(M
w
= 3870, M
w
/M
n
=
1.06, radius of gyration R
g
=19.2
˚
A) at 470 K and (b) a PDMS melt (M
w
= 6460,
M
w
/M
n
=1.10, R
g
=21.3
˚
A) at 373 K. The Q-values (in
˚
A
−1
)arenotedinthe
figures. The solid lines are the respective predictions of the Rouse model.
going to shorter length scales this condition must break down. Measurements
of the dynamic structure factor at smaller scales or higher Q-values offer the
opportunity to learn about the limitation of the Rouse model towards local
scales and to understand the leading processes. Both local chain stiffness as
well as rotational potentials may play a role [49–51].
Recently, the relevant factors, which limit the universal Rouse dynamics
towards small scales, were studied in comparing the dynamic structure factors
S(Q, t) of polyisobutylene (PIB) and polydimethylsiloxane (PDMS) of simi-
lar size in the melt and in dilute solution [21,22]. Both polymers show similar
static flexibility (the characteristic ratios are very close), but strongly differ-
ent rotational barriers (PDMS: E
rot
∼
=0.4kJ/mol; PIB: E
rot
=12.6kJ/mol).
542 Dieter Richter
Figure 13.22a presents the dynamic structure factor from a PIB melt at
470 K including a fit with the prediction of the Rouse model. Figure 13.22b
displays comparable data from a PDMS melt at 373 K.
While for Q-values above Q =0.15
˚
A
−1
increasing deviations between
the experimental results from PIB and the Rouse prediction are evident, the
PDMS results are described well over the entire Q-range (up to Q =0.4
˚
A
−1
).
Since both polymers exhibit the same static flexibility, we may immediately
conclude, that chain stiffness is not the leading mechanism limiting the Rouse
dynamics for flexible polymers as proposed earlier [51]. A thorough study of
the stiffness effects on the dynamics of this polymer was carried out in [21]
applying the approaches of the all-rotational-state model and a bending force
model. Stiffness effects were found to be almost negligible.
After the failure of stiffness models, the source for the slowing down of
the PIB chain dynamics at intermediate scales must be related to dissipation
effects not present in PDMS. Allegra’s intrachain viscosity model [49,50] pro-
vides a simple access to such an effect. He describes the intrachain dissipation
degrees of freedom by a local relaxation mode characterized by a relaxation
time τ
0
.
In order to access this process without disturbances experiments on so-
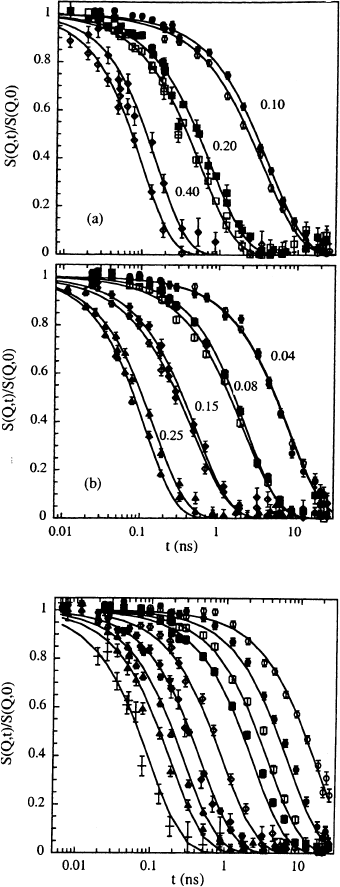
lutions were crucial, where interchain friction effects are weak [22]. Fig-
ures 13.23a,b compare experimental results on PIB and PDMS in toluene
at room temperature and 378 K. While at low Q both data sets agree – both
polymers undergo the same translational diffusion – at larger Q a systematic
retardation of the dynamic response of PIB compared to PDMS is visible.
The application of the intrachain viscosity model to the PIB solution data
on the basis of the PDMS reference led to a very good description of the NSE
results for all Q values and temperatures (Fig. 13.24). The activation energy
of 13 kJ/mol for the intrachain relaxation time τ
0
agrees very well with the
rotational barrier for this hydrocarbon [52].
An application of the model to the melt data allows a quantitative de-
scription of the Q-dependent spectra. The activation energy for τ
0
resulted
to be about 40 kJ/mol, i.e. more than 3 times as high as in solution. Ob-
viously, chain relaxation occurs via correlated motions over several barriers
or interchain effects come in additionally and the activation energy cannot
easily be compared with rotational potentials, see also [35].
Drawing a fourth conclusion, we note that Allegra’s intrachain viscosity
model accounts properly for the deviations from universal dynamics towards
shorter time and length scales. In solution, the corresponding relaxation time
directly relates to jumps across rotational potentials. Also in the melt, intra-
chain friction effects limit the Rouse dynamics. The relaxation time, however,
is not directly related to crossings of single barriers.
13 Viscoelasticity and Microscopic Motion in Dense Polymer Systems 543
Fig. 13.23. Chain-dynamic struc-
ture factor of PDMS (empty sym-
bols) and PIB (full symbols –
in each case the upper curve) in
toluene solution at 300 K (a) and
378 K (b). The corresponding Q (in
˚
A
−1
) values are indicated. Lines
through the points are guides to
the eye.
Fig. 13.24. Chain-dynamic struc-
ture factor of PIB in toluene so-
lution at 327 K at the Q values:
0.04
˚
A
−1
(◦); 0.06
˚
A
−1
(•); 0.08
˚
A
−1
(); 0.10
˚
A
−1
(); 0.15
˚
A
−1
(♦);
0.20
˚
A
−1
(); 0.25
˚
A
−1
(); 0.30
˚
A
−1
(); 0.40
˚
A
−1
(+). Solid lines cor-
respond to fitting curves with the
Rouse-Zimm model including intra-
chain viscosity.
13.7 The Crossover from Rouse to Reptation Dynamics
Chains of small molecular weight are not able to build entanglements and
their dynamics is supposed to be fully compatible with the Rouse model.
Increasing the chain length beyond a threshold length, topological hindrance
shows itself in (i) reducing the self-diffusion coefficient below the Rouse law
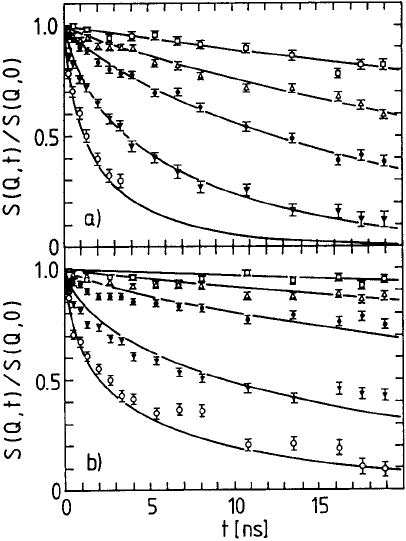
544 Dieter Richter
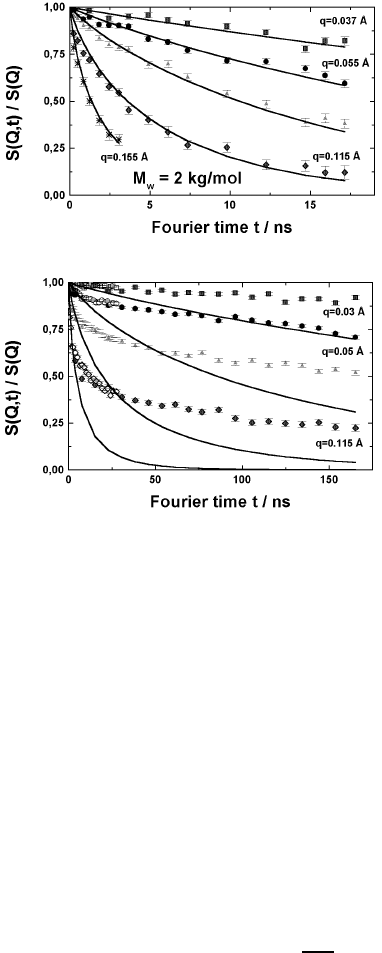
Fig. 13.25. Dynamic structure factor for two polyethylene melts of different mole-
cular weight; a) M
w
=2×10
3
b) M
w
=4.8 × 10
3
. The momentum transfers Q are
Q =0.037; 0.055; 0.077; 0.115 and 0.155
˚
A
−1
from top to bottom. The solid lines
show the result of mode analysis.
D
R
∼ 1/N and (ii) increasing the Rouse linear relationship between viscosity
and chain length to a power law η ∼ N
3.4
.
In order to find out how beginning entanglement effects influence the in-
trachain motions, we discuss neutron-spin echo experiments on a number of
polyethylene melts with molecular weights in the transition range (M
w
=
2000, 3600, 4800, 6500) [11, 13]. The monodisperse deuterated melts were
mixed with 10% protonated chains of the same length. Figure 13.25 shows
experimental spectra obtained on specimens with the molecular weights 2000
and 4800. A comparison of the spectra shows that the relaxation is obviously
much less advanced for the longer chain compared to the shorter chain. Let
us take the spectrum at the momentum transfer Q =0.12
˚
A
−1
as an ex-
ample. While the dynamic structure factor for the shorter chain has already
decreased to about 0.1 after 20 nanoseconds, the longer chain only relaxes to
about 0.4 for the same Q and the same time.
In the following analysis of these data we want to find out how the indi-
vidual normal modes of the relaxing chain are influenced by the appearance
13 Viscoelasticity and Microscopic Motion in Dense Polymer Systems 545
of topological hindrance. The starting point for this analysis is the fact that
the spatial structure of the eigenmodes of Gaussian chains is given by the
Rouse form
x
p
(t)=
1
N
n
x
n
(t)cos
pπn
N
. (13.23)
The time dependence can have an arbitrary form. Within the framework of
this mode structure, the dynamic structure factor can be formulated in a
general form [53].
S(Q, t)=
1
N
exp
−
Q
2
6
(x
0
(t) − x
0
(0))
2
×
mn
exp
−
1
6
Q
2
2
|m − n|−
2
3
R
2
E
π
2
Q
2
p
1
p
2
×
cos
pπm
N
cos
pπn
N
(1 −x
p
(t)x
p
(0))
(13.24)
where x
0
denotes the center of mass coordinates and the pointed brackets
denote the thermal average. The correlation function (x
0
(t) − x
0
(0))
2
de-
scribes the diffusive motion of the molecular center of gravity. The relaxation
dynamics of the internal modes is hidden in the exact time dependence of
the correlators x
p
(t)x
p
(0). They describe the time-dependent development
of the motion of a normal mode “p”. In the case of the entropy-determined
Rouse motion, the correlators have the form
x
p
(t)x
p
(0)e
−t/τ
p
,
1
6
[x
0
(t) − x
p
(0)]
2
=
k
B
T
Nζ
0
t = D
R
t (13.25)
where D
R
is the Rouse diffusion coefficient. The beginning spatial restrictions
should lead to a more complicated time dependence.
How can we hope to extract the contributions of the different normal
modes from the relaxation behaviour of the dynamic structure factor? The
capability of neutron scattering for directly observing molecular motions on
their natural time and length scale enables the determination of the mode
contributions to the relaxation of S(Q, t). Different relaxation modes influ-
ence the scattering function in different Q ranges. Since the dynamic structure
factor is not simply decomposed into a sum or a product of mode contribu-
tions, this Q-dependence is not easy to represent. In order to make the effects
more transparent, we consider the maximum possible contribution of a given
mode “p” to the relaxation of the dynamic structure factor. This maximum
contribution is reached when the correlator x
p
(t)x
p
(0) in (13.24) has fallen
to zero. In order to keep the picture simple, we retain all the other relaxation
modes: x
s
(t)x
s
(0) =1fors = p.
Under these conditions, (13.24) indicates the maximum extend to which a
particular mode p can reduce S(Q, t) as a function of the momentum transfer
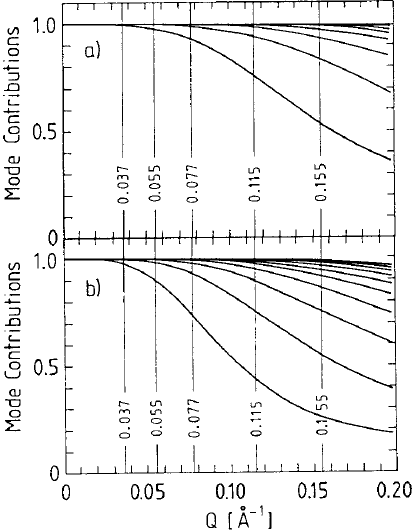
546 Dieter Richter
Fig. 13.26. Contributions of the different relaxation modes to the relaxation of
thedynamicstructurefactorS(Q, t)/S(Q, 0) for a) M
w
=2.0 × 10
3
and b) M
w
=
4.8×10
3
.TheexperimentalQ-values (in
˚
A
−1
) are indicated by vertical lines; curves
correspond to mode numbers increasing from bottom to top.
Q. Figure 13.26 presents the Q-dependence of the mode contributions for the
M
w
= 2000 and M
w
= 4800 samples. Vertical lines mark the experimentally
examined momentum transfers. Let us begin with the short chain. For the
smallest Q the internal modes do not influence the dynamic structure factor.
There, only the translation diffusion is observed. With increasing Q,firstof
all, the first mode begins to play a role. If Q is further increased, higher
relaxation modes above Q =0.1
˚
A
−1
also begin to influence the dynamic
structure factor. If the chain length is enlarged, the influence of the different
internal relaxation modes shifts to smaller momentum transfers.
This Q-dependent contribution of different relaxation modes to S(Q, t)
permits a separation of their influence on the dynamic structure factor. In
order to be able to evaluate the data with a reasonable number of parameters,
as a first approximation we assume that the exponential correlation of the
correlators (cf. (13.25)) is maintained. For further treatment reference is made
to [13].
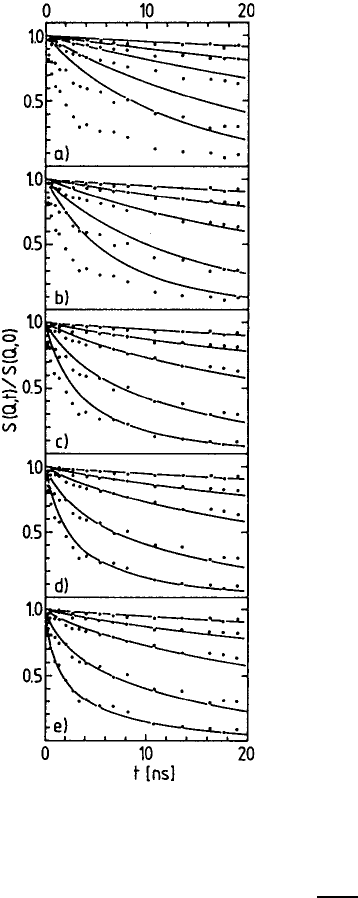
13 Viscoelasticity and Microscopic Motion in Dense Polymer Systems 547
Fig. 13.27. Resultofthemodeanalysisfor
the M
w
= 3600 sample. The diagrams show
the result of a calculation of the spectra re-
taining a successively rising number of modes
in comparison to the experimental result; a)
translation diffusion only, b) translation dif-
fusion and first mode, c) translation diffusion
and the first two modes, d) translation diffu-
sion and the first three modes, e) translation
diffusion and all modes.
The relaxation rates 1/τ
p
may depend in a general form on the mode
index p according to
1/τ
p
=
π
2
p
2
N
2
W
p
. (13.26)
The mode-dependent rate W
p
is the parameter of analysis.
Figure 13.27 demonstrates the contribution of different modes to the dy-
namic structure factor for the specimen with the molecular weight 3600.
Based on the parameters obtained in a common fit using (13.24) we cal-
culated S(Q, t) including an increasing number of mode contributions. Fig-
ures 13.27a show the contribution of translation diffusion. The translation
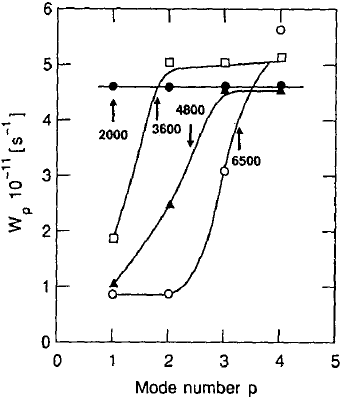
548 Dieter Richter
Fig. 13.28. Relaxation rates W
p
for the first four relaxation modes
of chains of different molecular
weight as a function of the mode
number p. The arrow indicates the
condition p = N/N
e
for each mole-
cular weight.
diffusion describes the experimental data for the smallest momentum trans-
fer Q =0.037
˚
A
−1
. Figure 13.27b presents S(Q, t) retaining the first mode.
Obviously, the long-time behaviour of the structure factor is now already
adequately represented, whereas at shorter times the chain apparently re-
laxes much faster than calculated. Figure 13.27c–e shows how the agreement
between experimental data and calculated structure factor improves if more
and more relaxation modes are included. In Fig. 13.27e, finally, very good
agreement between theory and experiment can be noted.
Figure 13.28 shows the results for W
p
, the mode-dependent relaxation
rate, for the different molecular weights as a function of the mode index
“p”. For the smallest molecular weight M
w
= 2000 relaxation rates W
p
are
obtained which are independent of p. This chain obviously follows the Rouse
law. The modes relax at a rate proportional to p
2
(cf. (13.15)). If the molecular
weight is increased, the relaxation rates are successively reduced for the low-
index modes in comparison to the Rouse relaxation, whereas the higher modes
remain uninfluenced within experimental error.
How can we understand this behaviour? From the long-chain results of
the last section we know that for polyethylene at the measuring temperature
of 509 K the molecular weight between the points of entanglement is about
M
e
= 2000 or the number of monomers between the entanglement points is
N
e
= M
e
/M
o
≈ 140 (M
o
: monomer weight). Let us assume that the charac-
teristic length for a relaxation mode L
p
is given by the distance between two
nodes (L
p
= N/p). We then may define a critical mode index p
cr
= N/N
e
below which the characteristic extension of a mode becomes greater than the
distance between entanglements in the long-chain melt. These critical mode
indices for the different molecular weights are plotted as arrows in Fig. 13.28.