Heitjans P., Karger J. (Eds.). Diffusion in Condensed Matter: Methods, Materials, Models
Подождите немного. Документ загружается.


1 Diffusion: Introduction and Case Studies in Metals and Binary Alloys 19
increased. Typical pressure effects range from factors of 2 to 10 for a pressure
of 1 GPa (10 kbar). This variation is largely due to the fact that the Gibbs
free energy of activation ∆G varies with pressure according to
∆G = ∆H − T∆S = ∆E − T∆S+ p∆V (1.23)
where ∆V denotes the activation volume and ∆E the activation energy of
diffusion. Using (1.21) and (1.23) the Arrhenius equation (1.20) can also be
written as
D = D
0
exp
−
∆G
RT
. (1.24)
Thermodynamics tells us that
∆V =
∂∆G
∂p
T
. (1.25)
Equation (1.25) can be considered as the definition of the activation volume.
Using (1.24) and (1.25) the activation volume can be obtained from mea-
surements of the diffusion coefficient at constant temperature as a function
of pressure via
∆V = −RT
∂ ln D
∂p
T
+ RT
∂ ln D
0
∂p
correction term
. (1.26)
The term arising from the pressure dependence of D
0
can be estimated from
the isothermal compressibility and the Grueneisen constant of the material.
It is often negligibly small (a few percent of the molar volume V
m
or of the
atomic volume Ω = V
m
/N
A
, respectively). Typical values for the activation
volume for various metals lie between 0.5 and 0.9 Ω. For a recent review
see [35].
We emphasize that a complete characterization of the diffusion process
requires knowledge of the three activation parameters ∆H = ∆E + p∆V
(valid for p =const.), ∆S and ∆V . For solids the term p∆V becomes signif-
icant only at high pressures. At ambient pressure it is negligible and hence
∆H ≈ ∆E.
1.6 Atomistic Description of Diffusion
1.6.1 Einstein-Smoluchowski Relation and Correlation Factor
Diffusion in crystalline solids takes place by a series of jumps of individual
atoms
7
. For interstitial diffusers the ‘diffusion lattice’ is formed by the in-
terstitial sites of the crystal lattice. Substitutional diffusers perform jumps
7
Near the melting temperature of fcc metals the self-diffusion coefficient (see
Sect. 1.8) has a value of about 10
−12
m
2
s
−1
. This corresponds to about 10 million
jumps of each atom per second.
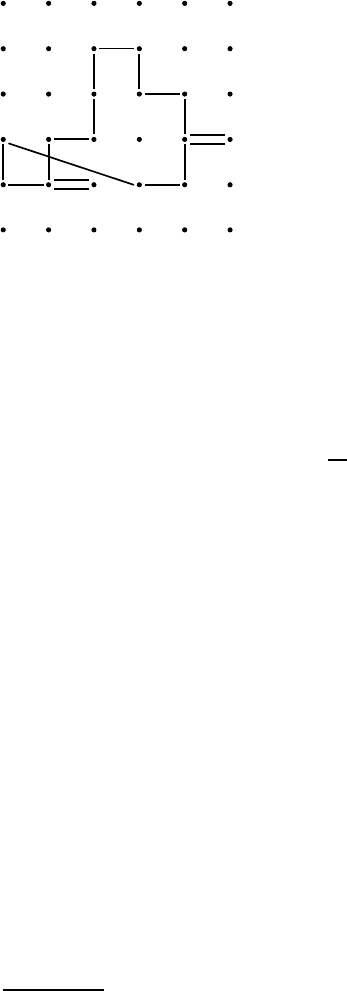
20 Helmut Mehrer
-
i
-
6
?
6
-
?
6
6
?
R
r
1
r
2
r
n
Fig. 1.6. Total displacement R of a particle on
a lattice composed of many individual jumps r
i
.
between sites of the normal lattice. The diffusivity on a lattice can be ex-
pressed in terms of physical quantities that describe the elementary jump
process. As we shall see below, these quantities are the jump length and the
jump rate.
The link between the displacement of particles on a (cubic) lattice and
the diffusion coefficient is given by the relation
8
D =
1
6t
R
2
(1.27)
where
R
2
denotes the mean square displacement of particles (see also
Chap. 18). Equation (1.27) is due to Einstein [36] and Smoluchowski [37].
It also includes correlation effects as we shall see below.
Suppose that a particle carries out n jumps during time t. As illustrated
in Fig. 1.6 its total displacement
R =
n
i=1
r
i
(1.28)
is composed of n individual jump vectors r
i
and hence
R
2
=
n
i=1
r
2
i
+2
n−1
i=1
n
j=i+1
r
i
r
j
. (1.29)
Taking the average over an ensemble of particles and using the fact that
the average of a sum of terms equals the sum of the averages of the terms we
get
R
2
=
n
i=1
r
2
i
+2
n−1
i=1
n
j=i+1
r
i
r
j
. (1.30)
8
We restrict ourselves for reasons of simplicity to cubic lattices. The extension to
non-cubic lattices is straightforward and can be found, e.g., in [2].

1 Diffusion: Introduction and Case Studies in Metals and Binary Alloys 21
Correlation effects are contained in the double sum of (1.30). For a true
random walk (index RW) the double sum in (1.30) vanishes as for every pair
r
i
r
j
one can find another pair equal and opposite in sign. Then
R
RW
2
=
n
i=1
r
2
i
. (1.31)
The degree of randomness in a long sequence of jumps can be expressed by
the so-called correlation factor f, which can be written as
f = lim
n→∞
R
2
R
2
RW
= 1 + 2 lim
n→∞
n−1
i=1
n
j=i+1
r
i
r
j
n
i=1
r
2
i
. (1.32)
For diffusion on a lattice the jump vectors r
i
can take only a few definite
values. Let us consider a cubic coordination lattice with coordination number
Z and let us suppose that only nearest-neighbour jumps of equal length l
occur. Then (1.31) reduces to
R
2
RW
=
n
l
2
, (1.33)
where
n
denotes the average number of jumps. For a true random walk (no
correlation) we get from (1.32) f = 1. It is convenient to define an average
jump rate of atoms into a certain direction, Γ ,by
Γ =
n
Zt
. (1.34)
The mean residence time τ of an atom on a certain lattice site is related to
the jump rate of the atom in a certain direction Γ via
τ =
1
ΓZ
. (1.35)
τ
−1
is the mean atomic jump rate in any direction on a lattice. Then (1.27)
can be written as
D =
1
6
l
2
ZΓ =
l
2
6τ
, (1.36)
which is identical with (1.14) discussed in Sect. 1.3. Diffusion of interstitial
atoms in a dilute interstitial solution can be considered as a true random
walk process and hence (1.36) is an appropriate expression for the diffusion
coefficient of interstitial diffusers.
Diffusion of self-atoms and of substitutional foreign atoms in metals,
metallic alloys, and in non-metallic solids is usually mediated by lattice va-
cancies. As already noticed by Bardeen and Herring [38] atoms migrating by
exchange with vacancies perform a correlated walk. The atom experiences
22 Helmut Mehrer
some ‘backward correlation’ since immediately after its exchange with the
vacancy the latter is still available for a backward hop of the atom. Then
the double sum in (1.30) does not vanish. For correlated diffusion on cubic
Bravais lattices
9
(1.36) must be replaced by
D =
1
6
fl
2
ZΓ. (1.37)
In summary we can say that the diffusion coefficient (in cubic crystals) for
interstitial diffusers as well as that for diffusers which migrate by exchange
with defects (vacancies, divacancies, self-interstitials, etc.) is described by
(1.37). For interstitial diffusers (in very dilute interstitial alloys) the corre-
lation factor is unity whereas for defect-mediated diffusion the correlation
factor is smaller than unity. Numerical values for the correlation factors of
self-diffusion can be found in Sect. 1.8. We emphasize that with increasing
degree of correlation the correlation factor decreases.
1.6.2 Atomic Jumps and Diffusion
Due to lattice vibrations atoms in a crystal oscillate around their equilibrium
positions with frequencies ν
0
of the order of the Debye frequency (typically
10
12
to 10
13
Hz). Usually an atom is confined to a certain site by a potential
barrier of height G
M
, which corresponds to the Gibbs free energy difference
between the configuration of the jumping atom at the saddle point and at
its equilibrium position. G
M
is denoted as Gibbs free energy of migration.
Diffusion is thermally activated which means that a fluctuation of thermal
energy pushes the atom over the energy barrier between two neighbouring
sites.
Using statistical thermodynamics Vineyard showed [39] that the jump
rate ω, at which an atomic jump into an empty neighbouring site occurs, can
bewrittenintheform
ω = ν
0
exp
−
G
M
RT
= ν
0
exp
S
M
R
exp
−
H
M
RT
. (1.38)
On the right-hand side of (1.38) the Gibbs free energy according to
G
M
= H
M
− TS
M
(1.39)
is decomposed into enthalpy H
M
and entropy S
M
of migration (superscript
M).
A diffusion jump of a self-atom or of a substitutional solute atom will only
occur if a neighbouring lattice site is occupied by a vacancy. If p denotes the
vacancy availability factor (the probability that the neighbouring site of the
9
For an extension to non-cubic lattices see, e.g., [2].
1 Diffusion: Introduction and Case Studies in Metals and Binary Alloys 23
jumping atom is empty) the jump rate of the atom to this particular site is
given by
Γ = ωp. (1.40)
Inserting this expression into (1.37) yields
D =
1
6
fl
2
Zωp. (1.41)
For interstitial diffusion in a very dilute interstitial alloy we have f =1,
p =1,andΓ ≡ ω. Then (1.41) reduces to (1.36).
1.6.3 Diffusion Mechanisms
Solid-state physics tells us that atoms in crystals oscillate around their equi-
librium positions. Occasionally these oscillations become large enough to al-
low an atom to change its site. As outlined above these jumps give rise to
diffusion in solids. Various atomic mechanisms of diffusion in crystals have
been identified and are catalogued in what follows:
Interstitial Mechanism
Solute atoms which are considerably smaller than the solvent (lattice)
atoms (e.g. hydrogen, carbon, nitrogen, and oxygen) are usually incorpo-
rated in interstitial sites of a metal. In this way an interstitial solid solution
is formed. Interstitial solutes usually occupy octahedral or tetrahedral sites
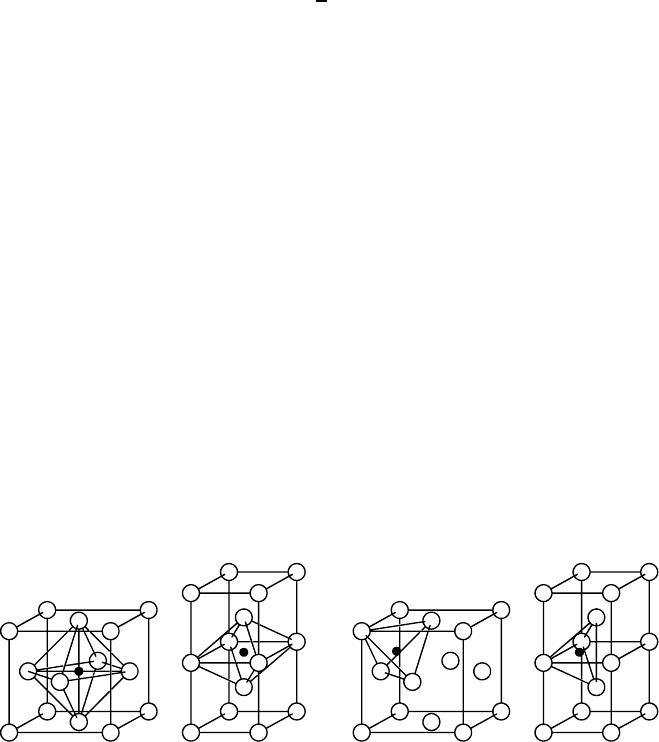
of the lattice. Octahedral and tetrahedral interstitial sites in the fcc and bcc
lattices are illustrated in Fig. 1.7. Interstitial solutes can diffuse by jumping
from one interstitial site to the next as shown in Fig. 1.8. This mechanism
is sometimes also denoted as direct interstitial mechanism in order to distin-
guish it more clearly from the interstitialcy mechanism discussed below.
(a) (b)
Fig. 1.7. Octahedral (a) and tetrahedral (b) interstitial sites in the fcc (left) and
bcc (right) lattice. Lattice atoms (open circles); interstitial sites (full circles).
Vacancy Mechanism
Self-atoms or substitutional solute atoms migrate by jumping into a neigh-
bouring vacant site as illustrated in Fig. 1.9. In thermal equilibrium the
atomic fraction of vacancies C
eq
V
in a monoatomic crystal is given by

24 Helmut Mehrer
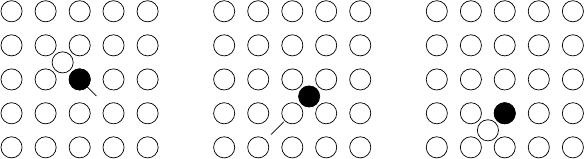
?
-
6
-
6
Fig. 1.8. Direct interstitial
mechanism of diffusion.
⇒
Fig. 1.9. Vacancy mechanism of diffu-
sion.
⇒
Fig. 1.10. Divacancy mechanism of dif-
fusion.
C
eq
V
=exp
S
F
R
exp
−
H
F
RT
, (1.42)
where S
F
and H
F
denote formation entropy and enthalpy of a vacancy (super-
script F ). For self-diffusion the vacancy availability factor p in (1.40) equals
C
eq
V
. Typical values for C
eq
V
near the melting temperature of metallic elements
lie between 10
−4
and 10
−3
(in molar fractions) [40]. From (1.38), (1.40), and
(1.42) we get for the jump rate of an atom in the case of self-diffusion
Γ = ν
0
exp
S
F
+ S
M
R
exp
−
H
F
+ H
M
RT
. (1.43)
Self-diffusion in metals and alloys, in many ionic crystals (e.g. alkali halides)
and ceramic materials occurs by the vacancy mechanism.
Attractive or repulsive interactions modify the probability to find a va-
cancy on a nearest-neighbour site of a solute atom. For a very dilute sub-
stitutional solution the probability p of finding a vacancy next to the solute
atom is given by
p = C
eq
V
exp
G
B
RT
, (1.44)
where G
B
denotes the Gibbs free energy of binding between vacancy and
solute (superscript B). The quantity G
F
−G
B
can be considered as the Gibbs
free energy for the formation of a vacancy on a nearest neighbour site of
the foreign atom. For attractive interaction (G
B
> 0) p is enhanced and for
repulsive interaction (G
B
< 0) p is reduced compared to the equilibrium site
fraction of vacancies C
eq
V
in a pure solvent
10
.
10
Note the convention: The binding energy is positive for attractive interaction.
1 Diffusion: Introduction and Case Studies in Metals and Binary Alloys 25
Divacancy Mechanism
Diffusion of self-atoms or substitutional solute atoms can also occur via
bound pairs of vacancies (denoted as divacancies or as vacancy pairs) as
illustrated in Fig. 1.10. At thermal equilibrium divacancies in an elemental
crystal are formed from monovacancies according to the reaction
V+V 2V . (1.45)
As a consequence of the law of mass action we have for the mole fractions
C
V
and C
2V
of mono- and divacancies
C
eq
2V
= K (C
eq
V
)
2
. (1.46)
The quantity K contains the Gibbs free binding energy of the vacancy pair.
Since the monovacancy population under equilibrium conditions increases
with temperature, the concentration of divacancies C
eq
2V
becomes more sig-
nificant at high temperatures. Divacancies in fcc metals have a higher mo-
bility than monovacancies [40]. Therefore self-diffusion of fcc metals usually
has some divacancy contribution in addition to the vacancy mechanism. The
latter is, however, the dominating mechanism at temperatures below 2/3of
the melting temperature [22].
Interstitialcy Mechanism
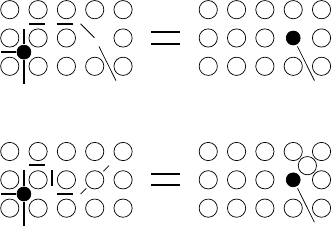
In this case self-interstitials – extra atoms located between lattice sites –
act as diffusion vehicles. As illustrated in Fig. 1.11 a self-interstitial replaces
an atom on a substitutional site which then replaces again a neighbouring
lattice atom. Self-interstitials are responsible for diffusion in the silver sublat-
tice of silver halides. In silicon, the base material of microelectronic devices,
the interstitialcy mechanism dominates self-diffusion and plays a prominent
role in the diffusion of some solute atoms including important doping ele-
ments [41]. This is not surprising since the diamond lattice (coordination
number 4) provides sufficient space for interstitial species. For a detailed dis-
cussion of the diffusion mechanism in Si see Chap. 4, Sect. 3.
In densely packed metals contributions of this mechanism are negligible
for thermal diffusion. Self-interstitials in metals have a fairly high formation
enthalpy compared to vacancies [40]. Therefore the concentration of the lat-
ter is dominating completely under equilibrium conditions. The interstitialcy
R
Fig. 1.11. Interstitialcy mechanism of diffusion.
26 Helmut Mehrer
mechanism is, however, important for radiation induced diffusion, since then
self-interstitials and vacancies are created in equal numbers by irradiation of
a crystal with energetic particles.
Interstitial-Substitutional Exchange Mechanisms
Some solute atoms (B) can be dissolved on interstitial (B
i
) and substi-
tutional sites (B
s
) of a solvent crystal (A) and diffuse via an interstitial-
substitutional exchange mechanism (see Fig. 1.12). For some of these so-called
‘hybride solutes’ the diffusivity of B
i
is much higher than the diffusivity of B
s
whereas the opposite is true for the solubilities. Under such conditions the
incorporation of B atoms can occur by the fast diffusion of B
i
and the sub-
sequent change-over to B
s
. Two types of interstitial-substitutional exchange
mechanisms can be distinguished:
If the change-over involves vacancies (V) according to
B
i
+V B
s
(1.47)
the mechanism is denoted as dissociative mechanism (sometimes also: Frank-
Turnbull mechanism or Longini mechanism). The rapid diffusion of Cu in
germanium (see Chap. 4, Sect. 4.3) and of some foreign metallic elements in
polyvalent metals such as lead, tin, niobium, titanium, and zirconium has
been attributed to this mechanism (see Sect. 1.9).
If the change-over involves self-interstitials (A
i
) according to
B
i
B
s
+A
i
(1.48)
the mechanism is denoted as kick-out mechanism. The fast diffusion of Au, Pt
and Zn in silicon has been attributed to this mechanism [41,42] (see Chap. 4,
Sect. 4.3).
For a description of diffusion processes, which involve interstitial-substitu-
tional exchange reactions, Fick’s equations must be supplemented by reaction
-
- -
6
R
-
B
i V V
-
-
6
?
-
-
B
i V
Fig. 1.12. Interstitial-substitutional exchange mechanisms of foreign atom diffu-
sion. Upper part: dissociative mechanism. Lower part: kick-out mechanism.

1 Diffusion: Introduction and Case Studies in Metals and Binary Alloys 27
terms. Since three species are involved, sets of three coupled diffusion-reaction
equations result for both mechanisms mentioned above. Solutions of these
equations – apart from a few (interesting) special cases – can only be ob-
tained by numerical methods. For details we refer the reader to the literature
(e.g. [41,42]).
In the following four sections we consider examples of normal interstitial
diffusion, diffusion of hydrogen, self-diffusion in pure metals, and of impurity
diffusion in metals
11
. We shall see below that the Arrhenius parameters of
(1.20) can be readily interpreted in terms of properties of atomic defects.
1.7 Interstitial Diffusion in Metals
1.7.1 ‘Normal’ Interstitial Solutes
Carbon, nitrogen, and oxygen often form interstitial solid solutions with met-
als. Interstitial sites in fcc and bcc lattices are illustrated in Fig. 1.7. Inter-
stitial diffusers have diffusion coefficients that are much larger than those
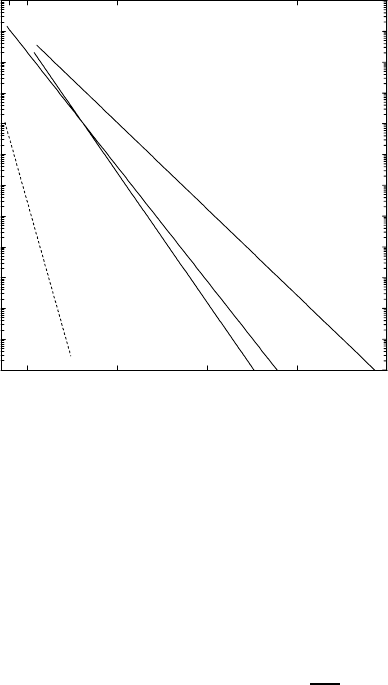
of self- and substitutional solute diffusion. Examples are shown in Fig. 1.13,
where diffusion of C, N, and O in niobium is displayed together with nio-
bium self-diffusion. The pertaining activation enthalpies and pre-exponential
factors are collected in Table 1.1 (for references see Chap. 8 in [6]).
Table 1.1. Activation parameters of some solutes in niobium (for references see [6]).
Diffuser C N O Nb
∆H [kJmol
−1
] 142 161 107 395
D
0
[m
2
s
−1
]1× 10
−6
6.3 × 10
−6
4.2 × 10
−7
5.2 × 10
−5
For a dilute interstitial alloy the probability p in (1.40) is unity. The
magnitude of the activation enthalpy is also small with the result that the
diffusion coefficients for interstitial diffusion are many orders of magnitude
larger than those for self-diffusion of lattice atoms. Interstitial diffusivities
near the melting temperature of the solvent can be as high as diffusivities in
liquids.
For interstitial solutes, which migrate by the direct interstitial mechanism,
(1.36) can be written as
D = ga
2
ν
0
exp
S
M
R
exp
−
H
M
RT
. (1.49)
Here a is the cubic lattice parameter and g a geometric factor, which depends
on the lattice geometry and on the type of interstitial sites (octahedral or
11
For reasons of simplicity we restrict ourselves to cubic crystals.
28 Helmut Mehrer
10
-20
10
-18
10
-16
10
-14
10
-12
10
-10
10
-8
5 10
15 20 25
2000 1000
500
O
C
N
N
b
T
-1
/ 10
-4
K
-1
D / m
2
s
-1
T / K
Fig. 1.13. Diffusion of C, N, O and Nb in niobium.
tetrahedral) occupied by the solute. For octahedral sites (Fig. 1.7) we have
g = 1 for the fcc lattice and g =1/6 for the bcc lattice. For interstitial
diffusers the Arrhenius parameters of (1.20) have the following meaning:
∆H = H
M
(1.50)
and
D
0
= ga
2
ν
0
exp
S
M
R
. (1.51)
With reasonable values for the migration entropy (between zero and several
R) and the attempt frequency (between 10
12
and 10
13
s
−1
) (1.51) yields the
following limits for the pre-exponential factors of interstitial solutes:
10
−7
m
2
s
−1
≤ D
0
≤ 10
−5
m
2
s
−1
. (1.52)
The experimental values of Table 1.1 confirm this estimate.
The fast diffusion of C, N, and O and the fact that N
2
and O
2
are gases, or,
in the case of carbon, readily available in gas or vapour form (CO
2
,CH
4
,...)
have an impact on the choice and availability of methods used to measure
their diffusion coefficients. Anelastic relaxation, magnetic relaxation (in the
case of ferromagnetic materials), internal friction, steady-state and in- and
out-diffusion methods are often applied. For details the reader is referred to
textbooks [2,3].