Heard D.E. (editor) Analytical Techniques for Atmospheric Measurement
Подождите немного. Документ загружается.


232 Analytical Techniques for Atmospheric Measurement
such as organic chemistry and physics. The first commercially available mass spectrometer
with magnetic sector mass filter was delivered in 1942 for the purpose of analysing
synthetically prepared organic compounds. Further developments quickly followed,
particularly in mass filters and detectors. The time of flight mass spectrometer (see
Section 5.3.2.2), quadrupole analyzer (Section 5.3.2.1) and the ion trap (Section 5.3.2.4)
were developed in the 1950s, and with the advent of the first computers systems became
more automated. The successful coupling of gas chromatography and mass spectrometry
improved the detection limits considerably, and as more commercial systems became
available in the 1960s, the technique proliferated throughout the field of science.
Mass spectrometers were first applied to atmospheric research in the 1960s. This work
was stimulated by the discovery that radio waves are reflected in the ionosphere and
a desire to understand the atmospheric distribution of ions. We now know that ions
are produced naturally in the atmosphere and their concentration controls the electrical
properties of the atmosphere as well as influencing important processes such as aerosol
production. Cosmic rays comprising extraterrestrial protons and alpha particles impinging
on the earth have sufficiently high energy to ionise N
2
(15.58 eV) and O
2
(12.07 eV).
Being charged, the resulting ions are affected by the earth’s magnetic field, analogously to
those in the mass spectrometer described above. The radiative emission of these particles
is the source of the Aurora seen at high latitudes. Over the first 100 km of the atmosphere
the ionisation density is relatively constant, around 10
3
ions cm
−3
. Since the neutral gas
density varies by a factor of 4×10
6
in this range, the mixing ratio of ions to neutrals
decreases substantially from high altitude to low altitude. At the surface there is only
one ion for circa 10
16
neutral molecules and the only significant source is from radon
(Viggiano & Arnold, 1995). Interestingly, for the reasons given above, the use of mass
spectrometry in the atmosphere began not at the ground but at high altitude (Narcisi &
Bailey, 1965). This was partly due to the interest in the ionosphere but mostly because
the high ion-to-neutrals ratio made it technically easier to make these measurements in
this region despite the inherent difficulty in getting the instruments to 60–100 km above
the Earth (see Section 5.2.1). Indeed it was only much later that the first measurements
of atmospheric ions were made from aircraft in the troposphere (Heitmann & Arnold,
1983), and then at ground level (Perkins & Eisele, 1984). Only positive atmospheric
ions were measured until 1971. In 1971, negative atmospheric ions were first measured
using rocket and balloon borne instruments (Arnold et al., 1971). This group discovered
that mass spectrometric measurements of naturally occurring ions in the middle to
lower atmosphere also provided information on the neutral trace gas composition (e.g.
acetone). It was a natural step to extend the measurement apparatus to include an
internal ion source, leading to the development of the atmospheric pressure ionisation
chemical ionisation mass spectrometer (API-CIMS) technique (see Section 5.4.1), based
on the principle of chemical ionisation first demonstrated by Munson and Field in 1966.
Through the 1980s these techniques permitted the measurement of important trace gases
in the upper troposphere and stratosphere. Airborne nitric acid measurements over the
Antarctic significantly contributed to the understanding of ozone hole chemistry. In the
laboratory, the improved sensitivity of continuous flow GC-MS allowed isotope ratios
of many important species to be determined (Barrie et al., 1984). In the 1990s chemical
ionisation mass spectrometry was developed for the measurement of important radical
species such as OH in the lower atmosphere. Despite its low concentrations and reactivity,

Mass Spectrometric Methods for Atmospheric Trace Gases 233
this species is now routinely measured at Hohenpeissenberg, Germany (see Figure 5.2).
The capillary GC-MS has become the most widespread MS technique in atmospheric
research, being particularly suited for measurement of organohalogens, CFCs, HCFCs
and alkanes. In 2000, the proton transfer reaction mass spectrometry technique became
commercially available and has proved popular for measurements of selected oxygenates
on short time scales (e.g. methanol). The number of atmospheric species measured by
mass spectrometry continues to increase, while detection limits, sampling time, size and
weight of instrumentation decreases. Within a century, mass spectrometry has developed
from a single laboratory experiment to become the most powerful and prolific technique
in atmospheric trace gas research at the time of writing, being applied from the tropics
to the poles and from the ground to the top of the atmosphere. Several examples of the
application of mass spectrometry in atmospheric field studies are given in Figure 5.2.
Figure 5.2 Examples of where trace gas mass spectrometry is used in atmospheric science.
(1) Ground-based APICIMS at Hohenpeissenberg for the routine measurement of OH. http://www.dwd.
de/en/FundE/Observator/MOHP/MOHP.html.
(2) Balloon launch of mass spectrometer to stratosphere from Kiruna, Sweden (Arnold & Henschen, 1978).
(3) The German research ship Meteor carrying PTR-MS and GC-MS measurements (Williams et al., 2004).
(4) Aircraft flying PTR-MS over the Amazonian rainforest (Crutzen et al., 2000).
(5) Commercial passenger aircraft carrying mass spectrometers and taking air samples on a regular basis for
long-term monitoring of the atmosphere (http://www.caribic-atmospheric.com/).

234 Analytical Techniques for Atmospheric Measurement
5.2 Fundamentals
5.2.1 The importance of vacuum
In Section 5.1 we have described mass spectrometry as involving the selective manip-
ulation of ions to a detector by electric and magnetic fields. Accurate deflection of
ions can only be achieved at low pressures as otherwise frequent collisions with neutral
molecules would destroy the order in the trajectories and possibly produce unwanted
ion–molecule reactions. Thus vacuum is essential for mass spectrometric analysis. From
kinetic theory the mean free path (L in m) of an ion at room temperature and pressure
can be calculated by:
L =
kT
√
2 ×p ×
(5.1)
Where k is the Boltzman constant 1381×10
−23
JK
−1
, T is the temperature (in K), p is
the pressure (in Pa) and is the collision cross section (typically 50×10
−20
m
2
). For mass
spectrometry a mean free path of circa 1 m is required, generally somewhat longer than
the mass filter. It follows that the maximum pressure should be 66 ×10
−3
Pa (66 nbar).
Furthermore, where high voltages are used in ion sources or mass filters, the pressure
must be further reduced to prevent arcing.
For the reasons given above, the invention of reliable and effective pumping methods
was key to the development of mass spectrometry, see Table 5.1. This is normally done
in two steps. First, a mechanical pump is used to reduce the pressure from ambient
pressure 1 bar 10
5
Pa to around a millibar 10
2
Pa. Nowadays clean diaphragm pumps
are preferred in place of the older rotary oil sealed pumps. To achieve high vacuum
(better than a millibar) an alternative pumping principle to that of compression must be
used. For this a diffusion pump is the simplest form; however, in modern instruments
these are being superseded by turbo molecular pumps. The later consist of several stages
of fast rotating blades (typically 60 000 rpm) that drive molecules to a mechanical pump.
Since the molecular pumps cannot operate at near ambient pressure they must be used in
combination with a mechanical pre-pump. When used to analyse ambient air in the field,
care must be taken to ensure that pumps are not shaken or vibrated excessively. This is
particularly true for the fast-rotating turbo pumps. When operated on aircraft or ships,
pumps are often mounted on shock-absorbing mounts or the entire mass spectrometer
is switched off before landing to prevent damage.
5.2.2 The atomic mass scale
Atomic and molecular weights used in mass spectrometry are generally expressed in terms
of atomic mass units (amu) or daltons. This unit is based on a relative scale in which the
reference is the carbon isotope
12
C, which has been assigned the mass of exactly 12 amu.
Therefore, by definition, 1 amu is 1/12 of the mass of one neutral
12
C atom.
1 amu =1 dalton = 1/1212 g
12
C/mol
12
C/60221 ×10
23
atoms
12
C/mol
12
C
= 166054 ×10
−24
g/atom
12
C =166054 ×10
−27
kg/atom
12
C

Mass Spectrometric Methods for Atmospheric Trace Gases 235
The atomic weight of an isotope such as
35
Cl is related to the reference
12
C by comparing
the two masses. Since the chlorine isotope is 2.914707 times heavier than that of the
carbon isotope the mass of the chlorine isotope is:
35
Cl =120000 dalton ×219407 =349688 dalton
As 1 mol of
12
C weighs 12.0000 g, the atomic weight of
35
Cl is 34.9688 g/mol.
One result of this relative scale is that the masses of most ions in a mass spectrum fall
near but not at integer values. Acetone (propanone), for example, has an exact mass of
58.0418.
3 ×120000 +6 ×100783 +1 ×1599491
CHO
Table 5.2 gives the exact masses of a selection of isotopes of common elements and
selected atmospheric trace gas species.
Note that for stoichiometric calculations, chemists use the average mass calculated using
the atomic weight, which is an average of the isotopes of each element of the molecule. The
average mass of methane would therefore be 1201115 +4 ×100797=160434 daltons
(see Table 5.2). However, in mass spectrometry, the monoisotopic mass generally is
used. This mass is calculated by using the mass of the most abundant isotope for each
constituent element. Methane is therefore 120000 +4 ×100783 = 1603132 daltons. In
mass spectrometry we may need to distinguish between ions with almost identical masses.
The extent to which they can be differentiated depends on the resolution of the mass
Table 5.2 Exact masses of selected elements and molecules
Element Atomic mass/daltons
1
H 1.00783
2
H 2.01410
12
C 12.00000
13
C 13.00335
14
N 14.00307
15
N 15.00011
16
O 15.99491
18
O 17.99916
35
Cl 34.96590
37
Cl 36.96590
C
5
H
8
68.06264
C
4
H
5
N 67.04222 (Low resolution needed)
CH
3
COCH
3
58.0418
HC(O)CH(O) 58.00548 (Medium resolution needed)
12
CH
3
D 17.03758
13
CH
4
17.03466 (High resolution needed)

236 Analytical Techniques for Atmospheric Measurement
spectrometer. In Table 5.2 the masses are given with four or five figures to the right of
the decimal point. High resolution mass spectrometers have this precision.
As has been noted above, ions are required to perform mass spectrometry. How these
ions interact with applied electric and magnetic fields depends on the mass and the
charge of the ions. It is important to note that a mass spectrometer determines the
mass-to-charge ratio m/z of these ions, see Figure 5.1. For example, singly charged
methane
12
C
1
H
+
4
, has m/z = 1603132 whereas
13
CH
2+
4
has m/z =8518. Where z is the
total charge of the ion Q divided by the charge of one electron e:
z = Q/e e =16 ×10
−19
Coulomb (5.2)
If mass is given in daltons and charge is given in electron charges, m/z is given in
thomson (Th). In some cases the unitless terms mass number and charge number are
given, in which case m/z is also unitless. Most ions encountered in mass spectrometry
have a charge corresponding to the loss of one electron. For this reason the term ‘mass-
to-charge ratio’ is often shortened to mass.
5.2.3 Resolving power and resolution
The resolving power of a mass spectrometer is a measure of its ability to separate two
ions of any defined mass difference. The theoretical resolving power necessary to separate
two ions differing in their m/z is defined as M/M, where M is the mean of the two
masses and M is the difference between the two masses. In order to separate an ion
of the major biogenic emission product isoprene m/z 68 from an ion of the biomass
burning product pyrrole m/z 67 a theoretical resolution of about 68 is required. This
is considered to be low resolving power. A medium resolving power of circa 1600 is
required to resolve the atmospherically important molecule acetone m/z 5804186 from
the shorter-lived whole mass isomer glyoxal m/z 5800547 (see Table 5.2). Note that
the isomers acetone and propanal have exactly the same mass and therefore cannot be
resolved. Methane is an important trace gas in the atmosphere and valuable information
about its sources and sinks can be obtained by measuring its isotopes. This is complicated
as the methane isotopes
13
CH
4
and
12
CH
3
D require a mass spectrometer with higher
resolving power, in this case ca. 5800. The original mass spectrometer built by Thomson
had a resolving power of about 13 which was improved tenfold by his student Aston in
1933, see Table 5.1. Commercial mass spectrometers are available with resolving powers
from a few hundred to 500 000.
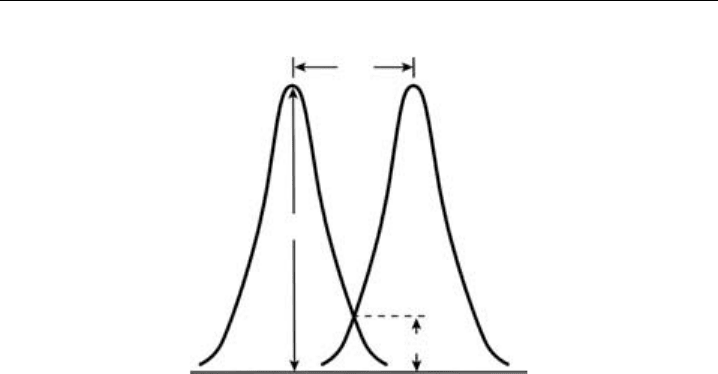
In Figure 5.3, two overlapping mass peaks M
1
and M
2
in a mass spectrum are depicted.
The resolving powers calculated above do not specify the acceptable degree of separation
between the ions with slightly different masses. This is given by the resolution R, which is
defined as the smallest mass difference for which two masses (M
1
and M
2
) are resolved. By
general agreement the peaks M
1
and M
2
are said to be resolved if the height of the valley
between the peaks h is equal or less than 10% of the peak height H. The resolution R
therefore defines the smallest mass difference M of peak width for which the two peaks
M
1
and M
2
are resolved to this extent. Many quadrupole instruments operate at constant
Mass Spectrometric Methods for Atmospheric Trace Gases 237
h
H
M
1
M
2
ΔM
Figure 5.3 Parameters defining peak resolution.
resolution. This means that the ability to separate mass 100 and mass 1000 is the same.
Note that if the resolution M is 1 amu at m/z =100, then the resolving power is 100.
However if M is 1 amu at m/z = 1000, then the resolution is still one but the resolving
power is 1000. In practice, the resolution may be set by introducing compounds at a
suitable mixture into the mass spectrometer to generate peaks M
1
and M
2
. Adjustment of
the various operating parameters of the mass spectrometer is performed until the peaks
have the desired resolution, i.e., 10% valley. Since sometimes it is difficult to get two
mass spectral peaks of equal height adjacent to one another, an alternative method of
calculating M is to use the full width at half maximum (FWHM) of a single m/z value
peak. Resolving power calculated using the FWHM method gives values for R that are
about twice that determined by the 10% valley method. By determining the resolving
power by using the full width at 5% full maximum, an R value close to the 10% valley
method can be derived. However, this is very difficult in practice because of background
signals.
5.3 Key elements of a mass spectrometer
5.3.1 Ionisation
The first step in the mass spectrometric analysis of an air sample is the ionisation of the
sample. The method used to ionise a substance profoundly influences the appearance of
its mass spectrum. For trace gas species in ambient air, two methods of ionisation are
currently prevalent. These are termed ‘electron ionisation’ and ‘chemical ionisation’ and
discussed in Sections 5.3.1.1 and 5.3.1.2 respectively.

238 Analytical Techniques for Atmospheric Measurement
5.3.1.1 Electron ionisation
Electrons produced from a heated filament in an evacuated chamber can be accelerated
towards an anode by applying a voltage (V). The energy of these electrons will be eV,
where e is the charge of the electron. Each electron is associated with a wave whose
wavelength ,
=
h
mv
(5.3)
where m is its mass, v is its velocity and h is Planck’s constant. For kinetic energies of
70 eV the wavelength is 14 ×10
−10
m. When this wavelength is close to the bond lengths,
the electron waves and the electric field of the molecule mutually distort one another. The
distorted electron wave can be considered to be many waves with different frequencies
and if one of the waves has the correct frequency (or energy) to interact with molecular
electrons (i.e. the energy corresponding to a transition), an energy transfer can occur. The
interaction of an electron with a molecule is rather non-specific. One possibility is that
the molecule may be electronically excited, in similar fashion to ultraviolet spectroscopy
(see Chapter 3). This can be represented by
M +e →M
∗
+e
−
(5.4)
Alternatively a molecular electron may be ejected from the molecule to leave a radical
cation. This is the best outcome for subsequent mass spectrometry since it produces an
ion that can be manipulated in electric and magnetic fields.
M +e
−
→ M
+
·+2e
−
(5.5)
A further possibility is direct capture of the electron by the molecule to give a stable
radical anion. However, the probability of this is low. Although less than one per cent
of the sample molecules are converted to positive ions, electron capture as depicted in
Equation (5.6) is probably one hundred times less probable.
M +e
−
→ M
−
· (5.6)
The energy of the ionising electrons may be varied by varying the applied voltage.
By convention, standard mass spectra are obtained at 70 eV since the maximum ion
yield is obtained near this energy. Furthermore, at this level small changes in energy
do not greatly affect the mass spectrum, helping the reproducibility of the analysis.
Mass spectra can be obtained at any electron energy above the ionisation energy of the
molecule (typically between 6 and 12 eV). Indeed it is often desirable to measure the
mass spectrum at energy levels less than 70 eV, despite the reduction in ion abundance,
since the amount of molecular fragmentation following ionisation can be reduced and
therefore identification facilitated. At higher potentials the electron wavelengths become
too small and molecules become effectively transparent. Extensive mass spectral libraries

Mass Spectrometric Methods for Atmospheric Trace Gases 239
have been compiled for substances ionised by electron ionisation at 70 eV. These can be
matched against unknown spectra from a sample to identify components.
5.3.1.2 Chemical ionisation
In chemical ionisation, ions are produced through the collision of the molecule to be
analysed with primary ions present in the source. The indirect method of chemical
ionisation (CI) is a complementary technique to the direct method of electron ionisation
(EI). While the high energies involved in EI commonly lead to fragmentation of the
molecular ion, chemical ionisation produces ions with little excess energy. Less fragmen-
tation facilitates identification of molecular species. Ion sources for chemical ionisation
are very similar to those for EI but they typically operate at high pressures (e.g. 0.1 mbar
compared to 10
−6
mbar for EI). In other words, in EI the gas sample is reduced in
pressure before the ionisation directly before the mass filter, while in CI the ionisation
generally occurs at higher pressure but the resulting ions must be directed via ion lenses
to the low pressure region containing the mass filter. In CI, both positive ions (e.g.
H
3
O
+
ions) and negative (e.g. SF
−
6
ions) can be used. Reactant gas ions are created by
leaking a reactant gas into an ion source where they interact with an electron beam or
high energy particles (e.g. emitted by radioactive sources) to produce primary ions. The
pressure in the ionisation region is adjusted so that the concentration ratio of reagent
to sample is 10
3
–10
4
. Because of this large concentration difference the electron interacts
predominantly with the reagent. The reagent ions then react with the sample molecules
of interest to produce ions. This form of chemical ionisation has two advantages over
electron ionisation described in Section 5.3.1.1. First, electron ionisation of a molecule
can give rise to a multitude of fragments, and for a complex mixture like ambient
air, the resulting mass spectrum can be indecipherable. Secondly by judiciously choosing
the reagent ion the trace gases of interest in the air can be detected preferentially over
the hugely more abundant species such as nitrogen and oxygen. Typical reagents are
methane, water and ammonia. Water is used for measurement of species such as acetone
by API-CIMS and PTR-MS (Sections 5.4.1 and 5.4.2). Methane has been used in negative
ion chemical ionisation in combination with gas chromatography, for the analysis of alkyl
nitrates and organohalogens in air (Section 5.4.4).
5.3.2 Mass filters
The mass filter is the heart of the mass spectrometer. It is here that the ions that have
been produced in the ion source are separated according to their m/z. The ability to
separate ions is characterised by the resolution (see Section 5.2.3). From the geometrical
configuration of the mass filters described below a theoretical resolution can be calculated.
However, in all cases the actual resolution is dependent on many other factors specific to
the device in question (e.g. constancy of the electric fields etc.) and should be empirically
assessed. The mass filters described below are all used in atmospheric research. In this
section the principle of each is described and in Sections 5.4 and 5.5 various applications
of these filters are described.
240 Analytical Techniques for Atmospheric Measurement
5.3.2.1 Quadrupole
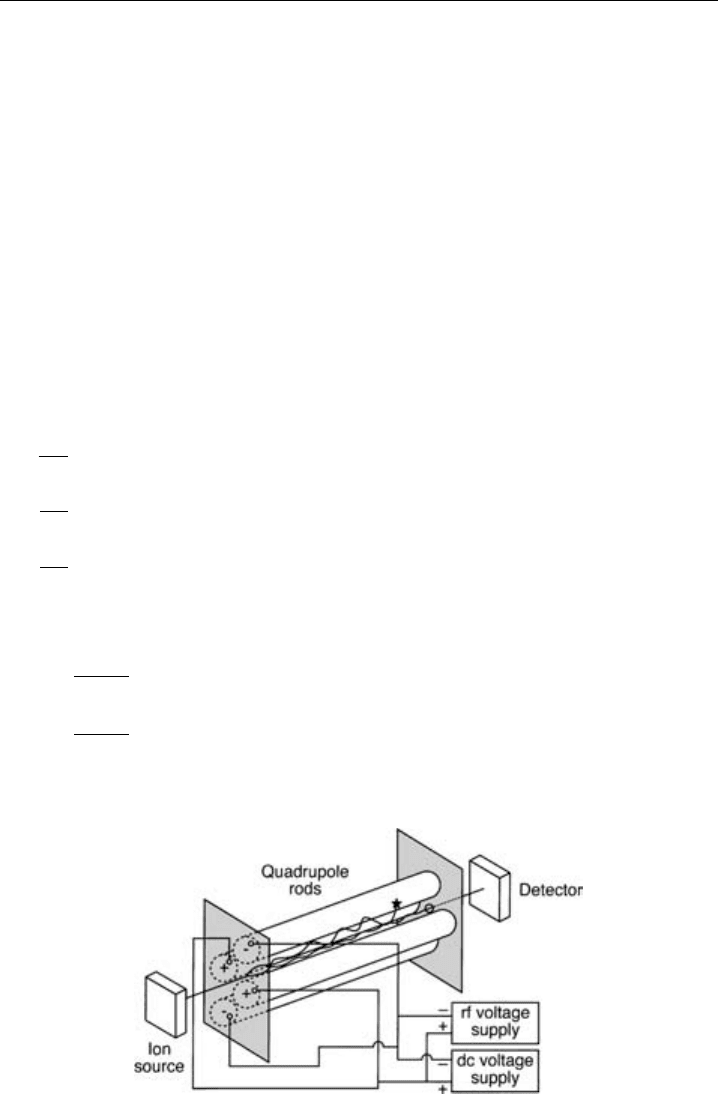
The quadrupole mass filter is a device that uses the stability of the trajectories in oscillating
electric fields to separate ions according to their m/z, see Figure 5.4. An ideal quadrupole
consists of four symmetrically arranged hyperbolic shaped surfaces of infinite length. In
reality, quadrupole mass filters consist of four parallel cylindrical rods. Typically these
are 10–20 mm in radius and 20 cm long. Opposing rods are electrically connected and a
potential comprising a DC voltage U and an RF component V ·cos t is applied. For
example when a positive ion enters the space between the rods, the electric field will act
to draw it to a negative rod. If the rod potential changes sign before the ion discharges
on the rod, the ion will change direction. Since there is no field applied in the z axis the
ions continue with the same speed as they enter the quadrupole. The DC and RF fields
can be varied so that an ion of a specific m/z can pass through the rods while others
do not.
The equations of motion of an ion in a quadrupole can be represented by the Mathieu
differential equations:
d
2
x
dt
2
+ a +2q cos t =0 (5.7)
d
2
y
dt
2
− a +2q cos ty = 0 (5.8)
d
2
z
dt
2
= 0 (5.9)
where,
a =
8eU
mr
2
0
2
(5.10)
q =
4eV
mr
2
0
2
(5.11)
t = 2 (5.12)
Figure 5.4 A schematic diagram of a quadrupole mass filter.

Mass Spectrometric Methods for Atmospheric Trace Gases 241
The solutions to these differential equations lie in two classes, corresponding to either
‘stable’ or ‘unstable’ trajectories. When the rod potentials (U, V and/or ) are varied in
a suitable manner then an ion of selected m/z (and only this ratio) may pass through the
quadrupole to the detector. By adjusting these potentials with time to satisfy a small mass
and then sequentially the larger masses, a mass spectrum can be generated. Figure 5.5
shows a so-called stability diagram for a quadrupole. Within the area defined by the
shark’s fin triangle are values of a and q generating stable solutions. The operating line
of the quadrupole is defined by the ratio a/q = 2U/V . By increasing this ratio, the
region of stable solutions can be so restricted that only ions of a specific mass pass
through. In this way the resolution of the mass filter can be varied, i.e. higher the ratio
the higher the resolution. However, when too high a ratio is chosen, the operating line is
no longer secant of the stable area and no ions will pass at all. For a real quadrupole, the
efficiency of transmission of ions depends on the exact locations and directions of the
ions when entering the rod system. Ions entering the rod system inclined to the z-axis
may possibly not pass the filter despite having the correct m/z. In fact the transmission
in the mass filter (number of ions entering/number of ions exiting) is approximately
inversely proportional to the resolution. Both transmission and resolution can influence
the accuracy and precision of a measurement and a compromise must be made between
the two for optimum performance.
Quadrupole mass filters are often operated at unit mass resolution (i.e. 1 amu, low
resolution), in contrast to the magnetic mass filters described in Section 5.3.2.3. However,
quadrupoles are relatively cheap and easy to operate. This is because the necessary
electronics (precise RF generators) have become standard technology instruments.
5.3.2.2 Time of flight
When ions of different masses are produced at the same place with the same energy, and
then accelerated by a potential V over a distance d, they will arrive at different times. The
time of flight mass filter relies on an accurate measurement of these times to distinguish
ions of different masses. In reality, ions are usually continually produced but expelled
in bundles from an ion source by the transient application of a potential to the source
0.2
0.1
Stable region
Unstable
region
Unstable
region
a/q = 0.15
0.0
0.0 0.2 0.4
q
0.6 0.8 1.0
a
Figure 5.5 The stability diagram for a quadrupole.