Gupta D. (Ed.). Diffusion Processes in Advanced Technological Materials
Подождите немного. Документ загружается.


292 DIFFUSION PROCESSES IN ADVANCED TECHNOLOGICAL MATERIALS
plotted as in Fig. 6.3 (namely L
2
versus t), and extrapolate to some
finite value of t for L zero, do not in fact satisfy the conditions for
such a plot. Instead, if we want to extract proper values for long-range
diffusion constants, they should be plotted as L versus t
12
, because
extrapolation gives a positive value of L for t 0. The physical cause
of this apparent paradox is that, initially, long-range oxidation is pre-
ceded by a period of anomalously fast oxidation, the source of which is
usually not analyzed in detail.
[12]
There is little doubt, however, that this
complexity arises as follows: The usual analysis of phase formation
kinetics, as above, is based on the assumption that the reactants remain
in their equilibrium state. However, this assumption is invalid during
the initial stage of oxidation, which is driven by very high free-energy
changes. When the oxide becomes thick enough and the rates slow
enough, the reacting surfaces (interfaces) return to equilibrium.
Initially, however, the rate of oxidation is too high to allow the sub-
strate atoms to relax to equilibrium positions and states before being
consumed by the growing oxide. This results in supersaturation of
defects, mostly vacancies during metal oxidation, and vacancies as well
as interstitials during Si oxidation, that indeed manifest their presence
in different ways, for example, dislocation loops, inside the substrate
itself. During that period, the equilibrium values of ∆G are not correct.
Excess values lead to enhanced growth rates and to the necessity of
plotting the data as L versus t
12
.
6.2.4 Linear-Parabolic Kinetics: Sequential
Phase Growth, Grain Boundary
Versus Lattice Diffusion
If several phases are likely to grow, then Eqs. (12a) and (12b) must be
written as:
dL
1
dt a[L
1
(aK
1
)] b[L
2
(bK
2
)] (16a)
and
dL
2
dt d[L
2
(dK
2
)] g[L
1
(gK
1
)]. (16b)
If phase 1 forms first, the growth of phase 2, from Eq. (16b), requires that:
d[L
2
(dK
2
)] g[L
1
(gK
1
)], (17)
Ch_06.qxd 11/12/04 4:05 PM Page 292

REACTIVE PHASE FORMATION, D’HEURLE ET AL. 293
which, for L
2
0, yields:
L
1c
d (1K
2
1K
1
). (18)
Phase 2 cannot grow until the first phase has reached a critical thickness L
1c
,
which increases as d (proportional to D
1
∆G
1
) increases, and decreases as
the initial rate of growth of phase 2, K
2
, increases. (If that rate is infinite, as
in the case of pure diffusion control, then L
1
c 0.) It becomes zero already
if K
1
K
2
; namely, there is no critical thickness for second-phase growth if
the maximum growth rates of the two phases are equal, which is indeed as
anticipated. An increase in K
1
, like an increase in d, increases L
1c
.
The literature on silicide formation relating to the reactions of thin
metallic films deposited either on single-crystal or polycrystalline Si bears
ample witness to the fact that phases do indeed form sequentially.
[15, 16]
With a 200-nm-thick film, growth of the first phase consumes all of the
metal present before a second phase begins to grow, for example, Ni
2
Si or
Pt
2
Si, then NiSi and PtSi, implying that L
1c
is larger than or of the order
of 300 nm.
[17–19]
As was emphasized above, such evidence is no confirma-
tion of a true reaction rate limitation at the beginning, so we should not
argue too much that no apparent activation energies have been determined
for this initial stage of silicide growth. What is certain is that when plot-
ted as in Fig. 6.3, the data show time intercepts that come closer to the ori-
gin as the temperature increases. Thus, if we compare observations made
on thin films, at relatively low temperatures, with observations on bulk
specimens, usually examined on a larger scale and consequently at con-
siderably higher temperatures, in the latter case, the sequential formation
of phases is usually ignored. This is probably more the result of the mode
of observation than of a true difference in behaviors between thin film and
bulk samples, which indeed obey the same laws except in extremely rare
cases (generally with extremely thin layers, below 50 nm). With thin
films, the interfaces tend to be quite clean. Retardation of phase formation
in bulk samples because of interface impurities, such as oxide with Al or
Ti samples, is not discussed here.
In discussing diffusion, the question of whether we refer to lattice or
grain boundary diffusion has been eluded. That was true also when writ-
ing the corresponding equations, which remain the same except for a
change in the value of D. Thus from the kinetics alone, it is difficult to
decide whether growth is due to one or the other of these two diffusion
mechanisms, since if the grain size of the growing layer remains con-
stant, the time exponent 1/n of the growth law remains 12. This ceases
to be true, however, if grain boundary diffusion is dominant and the
grain size increases during compound growth; then 1/n becomes smaller
Ch_06.qxd 11/12/04 4:05 PM Page 293

294 DIFFUSION PROCESSES IN ADVANCED TECHNOLOGICAL MATERIALS
than 12 (because diffusion slows down as the grain size increases). An
example is found in Sn-Ni reactions (for soldering) where a time expo-
nent smaller than 12 for the growth of Ni
3
Sn
2
is attributed to grain
growth.
[20]
The exponent reduces to 13 if the grain size is proportional
to the thickness of the layer, and even to 14 for normal grain growth,
where the driving force for grain growth is inversely proportional to the
grain size.
6.2.5 First Phase Formed
Alot of ill-spent ink has been poured on the question of what is the first
phase to form. Although it would be prudent to avoid the question altogether
here, it is nearly impossible to do so. According to what has been written in
Sec. 6.2.4, when a new phase is formed from the reaction between two ele-
ments, it is generally true that the free energies of formation of the different
phases do not differ much from each other: factors of 2, 3, or perhaps 4, but
rarely more than that. Thus the competition between phases as to which
would grow first depends on the product D
i
∆G
i
. It would be dominated
by D
i
’s, which differ much more widely than ∆G
i
, so that the first phase to
grow would be the one with the highest D,
[21]
or to be more precise, the
highest value for D ∆G. An extension of that statement based on some
observations about the relative diffusion of the two atomic species in binary
compounds, somewhat systematized as the “ordered Cu
3
Au rule,” predicts
that when there is a strong difference in melting points between the reac-
tants A and B, the first phase to form would be a phase rich in the element
with the low melting point, probably the one richest in that element.
[22]
A
more sophisticated approach,
[23]
based on irreversible thermodynamics,
implies that the first phase to grow should be the one with the highest rate
of free energy dissipation, namely, D ∆G
2
. However, inasmuch as the
∆G’s vary little from phase to phase in the initial reaction, the phase with
the highest D ∆G
2
product is likely to be that with the highest D.
Regarding the scale at which observations are made, the observations above
would seem to be valid for layers about 20 nm thick. At a smaller scale, we
might need to look into nucleation phenomena, about which little has been
said until now. With silicides, one can examine the process of formation of
amorphous phases reviewed by Sinclair.
[24]
6.2.6 Nucleation of the First or Second Phases
In Sec. 6.2.6, the description of phenomena that occur during what
can loosely be called the incubation time has remained deliberately vague,
Ch_06.qxd 11/12/04 4:05 PM Page 294
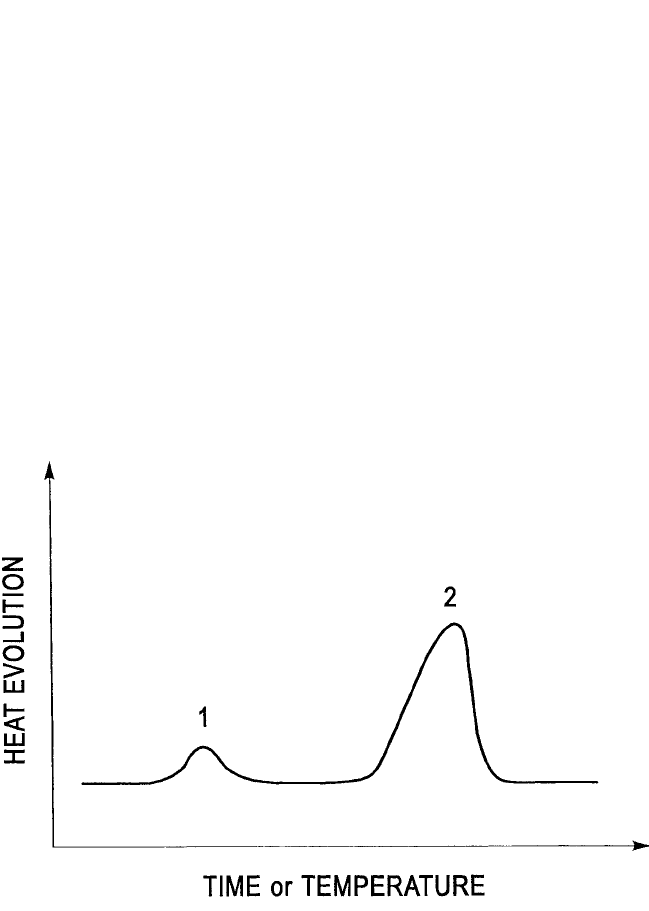
REACTIVE PHASE FORMATION, D’HEURLE ET AL. 295
except that the overall kinetics may be described as proportional to t
n
, with
n usually chosen for the sake of simplicity as 1, regardless of what is phys-
ically happening during this period. Considerable progress was made with
the discovery that the actual phase growth is preceded by an earlier
process related to nucleation;
[25]
the key to experimental success here was
the use of multilayered films that magnify the initial stage of a reaction in
comparison with the second stage, which is growth. During heating at a
constant heating rate, the evolution of heat occurs as shown in Fig. 6.4.
The first peak at low temperature results from the nucleation of the new
phase (in the case of silicides, often an amorphous one), which nucleates
first, then spreads along the original interface while maintaining a small
constant thickness. It is to be noted that the magnitude of the first exother-
mic peak in Fig. 6.4 is too large to be due entirely to nucleation, which
Figure 6.4 Heat evolution as a function of time during heating at a constant
rate of a reacting diffusion couple in a differential scanning calorimeter. There
are usually two peaks. The first, at low temperature, corresponds to a nucle-
ation step and often a lateral growth of the compound along the interface. The
second peak corresponds to what is usually considered growth itself. Very
interesting results have been obtained using multilayers that allow the relative
magnitudes of the two peaks to be affected almost at will. Does the picture
thus obtained provide a balanced view of what happens in an ordinary simple
diffusion couple?
Ch_06.qxd 11/12/04 4:05 PM Page 295

296 DIFFUSION PROCESSES IN ADVANCED TECHNOLOGICAL MATERIALS
contributes only a fraction of the total heat evolved in this first stage. Such
thermal behavior is not limited to silicides; it is also encountered with
other systems, such as Al-Co.
[26]
Although the literature should be studied
on a case-by-case basis, it is probably correct to say that we still need a
clear analysis of the transition between what occurs in stage 1 and the nor-
mal phase growth in phase 2, as described by Eqs. (4), (8), (12a), and
(12b).
Considerable work has been published regarding the nucleation that
occurs during peak 1 (Fig. 6.4). This research suffers from a number of
difficulties, many of which are common to all studies of nucleation phe-
nomena. The activation energy for nucleation ∆G* is the sum of two
terms:
∆G* ∆G*
th
∆G*
kin
, (19)
where ∆G*
kin
is the activation energy for the growth of a nucleus of criti-
cal size. In some cases, it may be equal or close to the activation energy
for growth that occurs later, but conceptually it is different. The other term
∆G*
th
has the form:
∆G*
th
→∆s
3
∆G
2
, (20)
where ∆s is the increase in interface energy on the formation of a nucleus
of critical size, and ∆G is the decrease in free energy (which should be
expressed here per unit volume) on the formation of the new phase. The
signs of ∆s and ∆G are opposite, and for small nuclei, the first term is
dominant. Thermodynamics dictate that the equilibrium density of nuclei
of critical size should be proportional to exp(∆G*
th
kT). The rate of
nucleation will necessarily be proportional to that quantity, yet nucleation
does not occur unless the critical nuclei grow. Therefore, as written in Eq.
(19), the activation energy for nucleation is the sum of two terms.
Diffusion theory states that the activation energy for diffusion via a
vacancy mechanism is also the sum of two terms: the energy for the for-
mation of a vacancy, plus the activation energy for the motion of these
vacancies. The case of nucleation considered here very closely parallels
diffusion.
Conceptually, the first term for the activation energy for nucleation
∆G*
th
is the easiest to manipulate, at least on paper. In principle, we know
what surface and interface energies are, so that we have an idea what ∆s
is. In principle also, we can assume that ∆G [in Eq. (20)] is the equilibrium
value for the formation of a new phase, as written in Eq. (1). Or, going
Ch_06.qxd 11/12/04 4:05 PM Page 296

REACTIVE PHASE FORMATION, D’HEURLE ET AL. 297
further, we can avail ourselves of more precise calculations of phase dia-
grams (CALPHAD) that will take into account the shape of the free energy
versus composition curves for the product and reactant phases. The situa-
tion for ∆G*
kin
is more ambiguous, because the formation of the nuclei may
occur along several competing paths with several different activation ener-
gies, especially when considering the formation of a compound new phase.
In fact, the conditions for any reliable calculations are often quite desper-
ate. Information about surface energies are usually lacking, and we are
unsure of the extent to which bulk or equilibrium free energies are valid for
submicroscopic particles. Nevertheless, it is quite current (although wrong)
to analyze activation energies for nucleation purely with respect to ∆G*
th
only, neglecting ∆G*
kin
, or assuming that in considering the rate of nucle-
ation, ∆G*
kin
is simply lost in the form of exp(∆G*
kin
kT) among the pre-
exponential terms in front of exp(∆G*
th
kT).
There may be some justification for neglecting the kinetic term for
nucleation, for example in the study of the precipitation of Co atoms in a
supersaturated solid solution of Co in Cu. That may occur via homoge-
neous or heterogeneous nucleation, and in comparing the two, we can
assume that ∆G*
kin
remains constant, so that experiments and calcula-
tions can be conducted with focus on ∆s (and therefore on ∆G*
th
) only.
When ∆G in Eq. (20) is very small, ∆G*
th
becomes very large, so that
qualitatively no harm is being done if ∆G*
kin
in Eq. (19) is neglected.
[27]
However, when ∆G*
kin
is neglected in attempting to determine which
phase forms first, for example, in the Ni-Al reaction,
[28]
which will be
used presently to illustrate difficulties encountered in the analysis of
competitive nucleation between phases of widely different compositions.
The Ni-Al system is almost ideal, because it is nearly completely sym-
metrical, with NiAl in the middle, Ni
3
Al and NiAl
3
on opposite sides of
the diagram, plus two intermediate phases, Ni
5
Al
3
and Ni
2
Al
3
. The
enthalpies of formation of the two extreme phases Ni
3
Al and NiAl
3
are
very close: 153 and 150 kJ/mol, respectively.
[29]
(This is surely
within the limits of accuracy of the measurements.) To demonstrate that
NiAl
3
should form preferentially to Ni
3
Al, the difference of enthalpies
between the terminal solid solutions is used: Al is somewhat soluble in
Ni; Ni in Al is not. It follows that the free energy of the Al in Ni solid
solution is somewhat lower than Ni in Al. Therefore, the ∆G (or ∆H) for
the formation of Ni
3
Al is somewhat smaller than ∆G for the formation of
NiAl
3
. This leads to a somewhat greater value for ∆G*
th
, and the pre-
ferred formation of NiAl
3
. However, considering that in one case the
phase is mostly composed of Ni and in the other mostly of Al, it is likely
that the differences in ∆G*
kin
would be 1 eV or more (because of the large
difference in melting points and consequent large difference in the
Ch_06.qxd 11/12/04 4:05 PM Page 297

298 DIFFUSION PROCESSES IN ADVANCED TECHNOLOGICAL MATERIALS
respective diffusion coefficients). Moreover, the enhanced thermody-
namic stability of a solid solution of Al in Ni, as opposed to that of Ni in
Al, implies that in the first case Al atoms will favor Al-Ni bonds, that is,
surround themselves with Ni atoms. Thus, since Ni
3
Al has the L1
2
struc-
ture, each Al atom in solid solution in Ni would constitute a potential
Ni
3
Al unitary cell. It is hard to understand that such a configuration
would not favor the nucleation of Ni
3
Al, independent of what the math-
ematics of ∆G*
th
imply.
We must also consider the fact that the nucleation of a new phase is
accompanied by sharp concentration and therefore also free energy gradients
because the very first phenomenon to occur is the superficial saturation of
A with B and B with A.
[30, 31]
Figure 6.5 shows that in such concentration
gradients, nucleation would happen preferentially in the phase, for exam-
ple, A, where the concentration gradient is minimum, because the diffu-
sion coefficient is maximum. Thus the phase to be nucleated would likely
be rich in A. From either Eq. (19) or Fig. 6.5, we conclude that the ques-
tion of the first phase to be nucleated is dominated by kinetic considera-
tions rather than by variations in ∆G*
th
. Moreover, if a phase with small
diffusion coefficients is nucleated first, it is likely to be immediately con-
sumed by another phase with faster kinetics, which has appropriately been
called vampire phase.
[32]
The discussion is carried out in terms of nucle-
ation,
[32]
but very much the same conclusions were reached earlier by sim-
ple considerations of competitive phase growth.
[33]
Thus it does not seem
that the analysis of nucleation is very helpful in understanding much of
the process of phase formation from reacting solid elements. Whether this
statement is generally valid or not, it is supported by a systematic analy-
sis of the reaction of Ti with Al, where experimental observations could
be understood in terms of competitive phase growth alone.
[34]
As antici-
pated, the first phase formed is rich in the element with the lowest melt-
ing point, Al.
Not much more can be said about nucleation in the present context.
It was emphasized above that whether we consider competitive phase
growth alone or nucleation in a concentration gradient (the usual condi-
tion), we are likely to predict that the first phase to grow will be rich in
the element with the lowest melting point. Further, in the reaction of Al
with Ni, NiAl
3
and Ni
2
Al
3
are rich in Al, so that in Eq. (19), the ∆G*
kin
for these two phases might be quite similar. Then in comparing NiAl
3
versus Ni
2
Al
3
, ∆G*
th
might play a significant role, quite different from
the role it would have in comparing NiAl
3
with Ni
3
Al. But in comparing
differences in ∆G*
th
, we are obliged to factor in the role of elastic energy
∆H
elas
, which can significantly affect the value of ∆G and about which
extremely little is known. In the absence of firm information about ∆s,
that term also remains somewhat unknown. We are left with experimental
Ch_06.qxd 11/12/04 4:05 PM Page 298
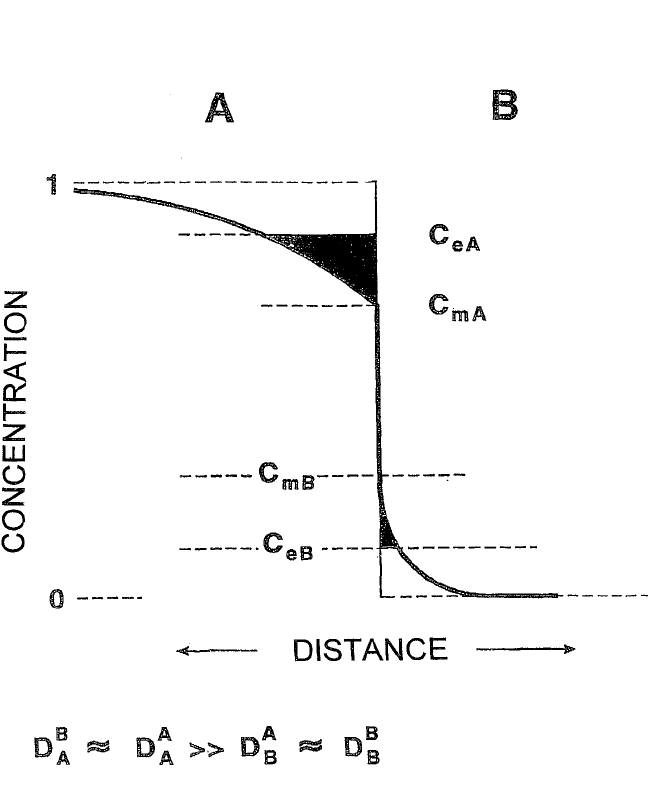
REACTIVE PHASE FORMATION, D’HEURLE ET AL. 299
Figure 6.5 Schematic representation of the initial step in a reaction between two
elements, A and B. The elements are assumed to have symmetrical thermody-
namic properties, as shown (the same mutual solubilities, c
eA
and c
eB
, at equilibrium,
or c
mA
and c
mB
, with the two elements in contact with each other), and similar com-
pounds (for example, A
3
B and AB
3
, not yet nucleated in the graph above), but dif-
ferent diffusion coefficients. (The diffusion coefficient of B in A, D
B
A
, is approxi-
mately equal to the coefficient of self-diffusion, D
A
A
. Both are much greater than
the corresponding coefficients in the other element, B.) The driving force G [Eq. (20)]
for the nucleation of an A-rich or B-rich compound is proportional to the blackened
areas on each side of the interface. Nucleation would then occur first on the side
of the equilibrium diagram where the element has a high diffusion coefficient. It fol-
lows that nucleation tends to be dominated by kinetic factors similar to those that
control growth. The reason for this is that, according to the Cu
3
Au rule,
[22]
if D
A
A
is
much greater than D
B
B
in general, D
A
A3B
would be much greater than D
B
AB3
, and
these latter coefficients control growth.
Ch_06.qxd 11/12/04 4:05 PM Page 299

300 DIFFUSION PROCESSES IN ADVANCED TECHNOLOGICAL MATERIALS
observations, which show that the reaction of Ti with Al produces
metastable forms of TiAl
3
with structures at variance with the equilib-
rium structure,
[35]
while Ni-Al reactions can also lead to a metastable
phase with a composition not encountered in the equilibrium diagram
Ni
2
Al
9
.
[36, 37]
(This appears to be in accordance with the Cu
3
Au rule
[22]
since, other things being equal, Al should be more mobile in Al
9
Ni
2
than
in Al
3
Ni.) Yet it is likely that nucleation processes play a role in deter-
mining which one of these phases is formed. However, conceptual
analysis has been of little help in resolving this problem. Small differ-
ences in sample preparation and heat treatment affect the observed
results, which does not simplify any realistic analysis. The complexity
is increased in a system such as Al-Pd, where many aluminum-rich com-
pounds are stable; reports of the first phase to form tend to change with
the investigators.
[38]
Whatever role nucleation plays in the selection of the formation of
these first or second phases, the relatively large ∆G in Eq. (1) [although
it is not the precise value that should be entered in Eq. (20)] results in a
relatively easy nucleation process and in a great abundance of nucle-
ation sites. Apractical effect of the numerous nucleation sites is that for
layers at least 10 nm thick, the interfaces are planar and parallel, as in Figs.
6.2 and 6.3. This is particularly noticeable with silicide reactions, where
the Si substrates are perfectly specular to begin with, and the samples
remain equally specular after the deposition of some 100 nm of Ni, as
well as after annealing to form Ni
2
Si and then NiSi. There are two gen-
eral aspects to be considered: (1) Reactive phase formation is usually
dominated by diffusion considerations, rather than nucleation. (2) In the
same way, kinetic factors, notably the growth modulated by diffusion,
dominate over thermodynamic ones. The fact that the first phase to grow
does not seem to depend significantly on the state of the reactants (solid,
liquid, or gas) provides experimental support for the first point. For
example, the disilicides NbSi
2
and MoSi
2
are obtained from the reaction
of thin metal films with Si substrates.
[39]
However, the same products are
obtained from the reaction of the metal with molten salt
[40]
or with SiH
4
(a gas),
[41]
respectively. The compound Cu
6
Sn
5
is obtained indifferently
whether Cu reacts with molten or solid Sn.
[20, 42, 43]
Although thermody-
namics is not unimportant, it indicates mostly what cannot happen: A
system cannot evolve in such a way as to increase the overall free energy
level. In the reaction of Mo with SiH
4
, MoSi
2
cannot be formed if the
pressure of SiH
4
and, consequently, the activity of Si are too low.
Gagliano et al.
[43]
provide a very good example of an experimental study
of nucleation in the reaction of Cu with liquid Sn. The potential role of
nucleation at the start of a reaction was theoretically investigated by
Gusak et al.
[44]
Ch_06.qxd 11/12/04 4:05 PM Page 300

REACTIVE PHASE FORMATION, D’HEURLE ET AL. 301
6.2.7 Nucleation-Controlled Reactions and
Consequences: Sequence of Phase
Formation, Bulk Samples, Stresses
If we follow the example of the formation of Ni silicides, we find
that with excess Si, the equilibrium phase should be NiSi
2
. However,
the formation of the equilibrium phase does not occur smoothly after
the formations of the two previous phases. These processes were dif-
fusion-controlled and occurred at about 350 and 400°C, respectively.
But once NiSi is formed, further heating does not cause any significant
change until a temperature above 750°C is reached. Then suddenly,
and apparently explosively, the sample transforms to NiSi
2
, with the
sample assuming a new frosted-glass appearance that is quite visible
to the naked eye. The nucleation of the new phase in this case was so
difficult that it required a relatively high temperature, where for sam-
ples of the order of 400 nm thick, diffusion was no longer rate-limit-
ing. The reason is simple: The overall ∆G (∆H) for the formation of
Ni
2
Si from Ni and Si is 143 ± 11 kJ/mol; that for the formation of
NiSi from the reaction of Ni
2
Si with Si is 18 ± 9 kJ/mol;
[27]
but for
the reaction:
NiSi Si NiSi
2
, (21)
the ∆H (∆G) is only 5 ± 20 kJ/mol. It is so small that it is not easily
accessible from experimental measurements. When the ∆H’s and ∆G’s
become small, it is no longer correct to consider that the two sets of val-
ues are identical.
[45]
However, what matters presently is that both values
are indeed quite small, so that from Eq. (20), ∆G*
th
becomes very large.
The practical consequences of the high temperature required for nucle-
ation to overcome this problem, the paucity of nuclei, and the rough sur-
face of the silicide layers are considered in Sec. 6.3. Even without knowing
the quantitative details of the nucleation process, games can be played:
Replacing monocrystalline Si with amorphous Si does not significantly
change the formations of either Ni
2
Si or NiSi, but the formation of NiSi
2
becomes diffusion-controlled.
[46]
The increase in ∆G, for Eq. (21), equal
to the difference in free energy between crystalline and amorphous Si, is
enough to cause a change in the mode of formation of NiSi
2
. Errors have
been made in interpreting reactions such as the formation of NiSi
2
, where
the sudden occurrence leads to the conclusion of a reactive explosion.
This is wrong. The nature of the reaction is dominated by a small ∆G and
∆H, and cannot therefore be truly explosive, especially on Si substrates
with a high coefficient of thermal conductivity, which limits any self-
Ch_06.qxd 11/12/04 4:05 PM Page 301