Faulon J.L., Bender A. Handbook of Chemoinformatics Algorithms
Подождите немного. Документ загружается.


408 Handbook of Chemoinformatics Algorithms
steps and the fate of carbon atoms within each step must be known a priori. The
system’s input and output fluxes are determined by quantitative physiological param-
eters obtained from the experiment, including nutrient feed, cell growth rates and
densities in the chemostat, and the efflux. The stoichiometric constraints on all inter-
nal fluxes are formulated using the principle of mass conservation, that is, N ·v = 0.
The isotopomer balance equations for the internal reactions are then derived using
either the BNGL/BioNetGen-based method or the IMM-based method. The under-
lying mathematical model for
13
C MFA is a set of isotopomer balance equations
as shown below in Equations 15.19 through 15.34. Given an initial guess for the
intermediate fluxes, the balance equations, in algebraic or ordinary differential equa-
tion (ODE) form, are used to compute the steady-state or dynamic labeling patterns.
The mathematical model for the dynamic
13
C labeling experiments is generally a
high-dimensional coupled system of differential algebraic equations, which in fact
constitutes an inverse problem for the unknown intracellular fluxes. Thus, predictions
for the internal fluxes are obtained by minimizing the difference between simulated
and measured isotope patterns. Essentially, this is a complex parameter fitting prob-
lem, which can be solved using a variety of techniques. For steady-state
13
C labeling
experiments, the model is a set of algebraic equations, and the algorithms employed
for numerical flux estimation are primarily gradient-based local optimization [27] or
gradient-free global optimization [10, 28–30] techniques, such as simulated anneal-
ing or genetic algorithms. In addition, a hybrid technique of global-local optimization
has been applied [31]. For dynamic
13
C labeling analysis, the system is described by
a set of algebraic–differential equations. Although analysis of dynamic
13
C label-
ing data has been proposed by a number of groups and used to estimate fluxes in
small metabolic subnetworks [32–34], to the best of our knowledge, this type of
analysis is yet to be used to estimate fluxes in large-scale networks. A barrier to
estimating large numbers of fluxes from this type of data is the large number of
isotopomer balance equations that must be specified. This problem is solved by the
BNGL/BioNetGen-based approach we review here.
Minimizing the objective function [31], that is, the difference between the sim-
ulated and measured labeling patterns using, for example, simulated annealing,
provides new estimates for the internal fluxes. The objective function is given by
Obj =
N
i=1
M
i
(t) −E
i
(
V, v, t)
δ
i
2
, (15.13)
where M
i
are the N individual labeling measurements and E
i
their corresponding
simulated values. The quantity δ
i
is the confidence value of the ith measurement.
For dynamic isotopomer experiments, M
i
is a function of time, and E
i
is a function
of time, V represents metabolite pool sizes, and v represents flux distributions. For
steady-state isotopomer experiments, E
i
is a function of flux distributions (v) only.
Using these revised fluxes, new isotope labeling patterns are generated and again
compared with the observed labeling patterns. This process continues until some
measure of convergence is achieved, for example, the intracellular fluxes do not
Using Systems Biology Techniques to Determine Metabolic Fluxes 409
significantly change between iterations or some predefined number of iterations is
performed.
15.3.2 IMM-BASED APPROACH
We now apply the IMM method to the four-flux problem posed in Figure 15.3 to
determine the metabolic fluxes. For a metabolite S with two carbons, the IDV is
given by
I
S
=
⎛
⎜
⎜
⎜
⎝
I
S
(00)
I
S
(01)
I
S
(10)
I
S
(11)
⎞
⎟
⎟
⎟
⎠
, (15.14)
where the binary representation (·) corresponds to the state (C1 C2), and 0 and 1
indicate
12
C and
13
C, respectively. 0 ≤ I
S
(j) ≤ 1 is the fraction of S molecules that
show a labeling pattern corresponding to the binary representation of j. We can derive
the IMM for each of the internal reactions directly from the carbon atom fate map
provided in Figure 15.3. IMM
S>M1
denotes the IMM for the reaction between S and
M1. We obtain
IMM
S>M1
=
⎛
⎜
⎜
⎜
⎝
1000
0100
0010
0001
⎞
⎟
⎟
⎟
⎠
, (15.15)
IMM
S>M2
=
⎛
⎜
⎜
⎜
⎝
1000
0010
0100
0001
⎞
⎟
⎟
⎟
⎠
, (15.16)
IMM
M1>P
=
⎛
⎜
⎜
⎜
⎝
1000
0100
0010
0001
⎞
⎟
⎟
⎟
⎠
, (15.17)
IMM
M2>P
=
⎛
⎜
⎜
⎜
⎝
1000
0100
0010
0001
⎞
⎟
⎟
⎟
⎠
. (15.18)
Each column of the IMM indicates how the substrate isotopomer evolves to the
corresponding isotopomers in the product, which is represented by each row. For
example, in IMM
S>M1
, the first column indicates that the isotopomer S
00
can only
be transferred to a product M1
00
.

410 Handbook of Chemoinformatics Algorithms
Again, for a reaction A→B, the isotopomer balance equations can be derived from
Equation 15.1. The corresponding isotopomer balance equations for our four-flux
example are therefore
V
S
dS
00
dt
= v
S
S
00
−(v
1
+v
2
)S
00
, (15.19)
V
S
dS
01
dt
= v
S
S
01
−(v
1
+v
2
)S
01
, (15.20)
V
S
dS
10
dt
= v
S
S
10
−(v
1
+v
2
)S
10
, (15.21)
V
S
dS
11
dt
= v
S
S
11
−(v
1
+v
2
)S
11
, (15.22)
V
M1
dM1
00
dt
= v
1
S
00
−(v
3
+v
M1
)M1
00
, (15.23)
V
M1
dM1
01
dt
= v
1
S
01
−(v
3
+v
M1
)M1
01
, (15.24)
V
M1
dM1
10
dt
= v
1
S
10
−(v
3
+v
M1
)M1
10
, (15.25)
V
M1
dM1
11
dt
= v
1
S
11
−(v
3
+v
M1
)M1
11
(15.26)
V
M2
dM2
00
dt
= v
2
S
00
−(v
4
+v
M2
)M2
00
, (15.27)
V
M2
dM2
01
dt
= v
2
S
01
−(v
4
+v
M2
)M2
01
, (15.28)
V
M2
dM2
10
dt
= v
2
S
10
−(v
4
+v
M2
)M2
10
, (15.29)
V
M2
dM2
11
dt
= v
2
S
11
−(v
4
+v
M2
)M2
11
, (15.30)
V
P
dP
00
dt
= v
3
M1
00
+v
4
M2
00
−v
P
P
00
, (15.31)
V
P
dP
01
dt
= v
3
M1
01
+v
4
M2
01
−v
P
P
01
, (15.32)
V
P
dP
10
dt
= v
3
M1
10
+v
4
M2
10
−v
P
P
10
(15.33)
V
P
dP
11
dt
= v
3
M1
11
+v
4
M2
11
−v
P
P
11
, (15.34)
and are subject to the following algebraic constraints:
S
00
+S
01
+S
10
+S
11
= 1, (15.35)
M1
00
+M1
01
+M1
10
+M1
11
= 1, (15.36)
Using Systems Biology Techniques to Determine Metabolic Fluxes 411
M2
00
+M2
01
+M2
10
+M2
11
= 1, (15.37)
P
00
+P
01
+P
10
+P
11
= 1. (15.38)
These equations arise due to the definition of the isotopomer fractions, that is, mass
conservation dictates that these fractions must sum to one. The isotopomer balance
equations coupled with the algebraic constraints comprise a system of differential–
algebraic equations that needs to be solved to determine the dynamic isotopomer
labeling patterns.
15.3.3 BNGL/BIONETGEN-BASED APPROACH
As discussed previously, BioNetGen derives the isotopomer balance equations rel-
evant for a given experiment directly from fate maps and generates the dynamic
distribution of observable metabolites given the input parameters. Here we simulate
the forward problem shown in Figure 15.3 using BioNetGen and generate the iso-
tope labeling patterns given the assumed flux patterns. The BioNetGen input file
for the four-flux example is provided below. It includes several blocks: parameters,
species, reaction rules, and observables. In the first block, the metabolite pool sizes
and the flux estimates, which are the input parameters, are specified. The second
block identifies the species, which are the metabolites, and their initial concentra-
tions. For each differently labeled initial metabolite, define a precursor species with
fixed concentration (defined by prepending the “$” character) corresponding to the
fraction in that labeling state. The block entitled “reaction rules” defines the carbon
atom fate maps. Finally, the observables, or model outputs, are given in the fourth
block. In this input file, the observables are the metabolite isotopomer fractions for
P. For example, the observable pattern P(C1∼1, C2∼0) represents the isotopomer
P
10
. The remaining sections are commands for generating the isotopomer balance
equations and simulating the dynamic labeling patterns. Specifically, the next block
of this input file contains commands that generate the detailed isotopomer reac-
tions from the specified reaction rules and write BioNetGen and MATLAB
®
output
files. The next several sections simulate the dynamic labeling patterns given differ-
ent pool sizes and/or fluxes. The command setParameter() defines the pool sizes
or the fluxes. The system is perturbed by feeding the system with a labeled sub-
strate. In this case, we have labeled 10% at the first carbon position. The command
simulate_ode() runs a simulation of the dynamic trajectories of the isotope labeling
patterns.
begin parameters
# Pools
V_S 1.0
V_M1 1.0
V_M2 1.0
V_P 1.0
412 Handbook of Chemoinformatics Algorithms
# Fluxes
v_v1 0.5
v_v2 0.5
v_v3 0.4
v_v4 0.4
v_vm1 0.1
v_vm2 0.1
v_vp 0.8
v_vs 1.0
# Labeled fraction
f_label 0.1
end parameters
begin species
$proS(C1 ∼0,C2 ∼0) 1-f_label
$proS(C1 ∼1,C2 ∼0) f_label
S(C1 ∼0,C2 ∼0) V_S
M1(C1 ∼0,C2 ∼0) V_M1
M2(C1 ∼0,C2 ∼0) V_M2
P(C1 ∼0,C2 ∼0) V_P
NULL 0
end parameters
begin reaction rules
pros(C1%1,C2%2) -> M1(C1%1,C2%2) v_v1/V_S
S(C1%1,C2%2) -> M1(C1%1,C2%2) v_v1/V_S
S(C1%1,C2%2) -> M2(C1%2,C2%1) v_v2/V_S
M1(C1%1,C2%2) -> P(C1%1,C2%2) v_v3/V_M1
M2(C1%1,C2%2) -> P(C1%1,C2%2) v_v4/V_M2
M1() -> NULL v_vm1/V_M1
M2() -> NULL v_vm2/V_M2
P() -> NULL v_vp/V_P
end reaction rules
begin observables
Molecules P00 P(C1 ∼0,C2 ∼0)/V_P
Molecules P01 P(C1 ∼0,C2 ∼1)/V_P
Molecules P10 P(C1 ∼1,C2 ∼0)/V_P
Molecules P11 P(C1 ∼1,C2 ∼1)/V_P
end observables
Using Systems Biology Techniques to Determine Metabolic Fluxes 413
generate_network({overwrite=>1});
writeSBML();
writeMfile();
saveConcentrations();
imulate_ode({t_end=>20, n_steps=>40});
resetConcentrations();
setParameter(V_yM2, 0.5);
simulate_yode({suffix=>"M2low", t_end=>20,
n_steps=>40});
resetConcentrations();
setParameter(V_yM2, 2.0);
simulate_ode({suffix=>"M2high", t_end=>20,
n{_}ysteps=>40});
resetConcentrations();
setParameter(V_M2, 1.0);
setParameter(v_v1, 0.6);
setParameter(v_v2, 0.4);
setParameter(v_v3, 0.5);
setParameter(v_v4, 0.3);
simulate_ode({suffix=>"dottedline", t_end=>20,
n_steps=>40});
The raw experimental measurements of isotopomer distributions contain the con-
tributions due to naturally occurring isotopes of its elements, such as hydrogen,
carbon, nitrogen, oxygen, and so on. Correction of the isotopomer distribution is
required to take into account differences in the relative natural abundance distribution
of each mass isotopomer (skewing) as described in Ref. [35]. A software tool for this
correction is reported in Ref. [36].
Solving the system of differential algebraic equations presented earlier gives us a
dynamic trajectory of labeling patterns. Figure 15.4 depicts the temporal dynamics
of the reaction product P
01
from a simulation of the four-flux network. P
01
indicates
that the second carbon is
13
C labeled. At time zero, the input feed changes from S
00
=
1.0, S
01
= S
10
= S
11
= 0toS
00
= 0.9, S
01
= 0, S
10
= 0.1, and S
11
= 0, indicating
that 10% of the feed is
13
C labeled at carbon position 1. We assume the following
values for the fluxes: v
S
= 1.0, v
1
= v
2
= 0.5, v
3
= v
4
= 0.4, v
M1
= v
M2
= 0.1,
and v
P
= 0.8. In Figure 15.4, we study the effect of metabolite pool size on product
dynamics. The pool sizes of the metabolites S, M1, and P are set to 1, and the pool size
of M2 is varied. We see that the pool size has no effect on the steady-state distribution.
Interestingly, however, Figure 15.4 shows that the dynamic labeling patterns depend,
not only on the fluxes, but also on the metabolite pool sizes. Consequently, valuable
information about the relative metabolite pool sizes can potentially be inferred from
plots of the temporal dynamics.

414 Handbook of Chemoinformatics Algorithms
0.05
0.04
0.03
0.02
P
01
0.01
0
0510
Time
15 20
FIGURE 15.4 Dynamic simulation of the four-flux problem. At time zero, the input feed
changes from S
00
= 1.0, S
01
= S
10
= S
11
= 0toS
00
= 0.9, S
01
= 0, S
10
= 0.1, and S
11
= 0.
Time evolution of the reaction product with
13
C at carbon position 2 (P
01
) for different pool
sizes is plotted. Flux patterns are assumed as follows: v
S
= 1.0, v
1
= v
2
= 0.5, v
3
= v
4
= 0.4,
v
M1
= v
M2
= 0.1, and v
P
= 0.8. The pool sizes of metabolites S, M1, and P are set to 1, and the
pool size of M2 is varied. M2 = 1 (solid line), M2 = 2 (dashed line), and M2 = 0.5 (dash-dot
line). For the dotted line, flux patterns are assumed as follows: v
S
= 1.0, v
1
= 0.6, v
2
= 0.4,
v
3
= 0.5, v
4
= 0.3, v
M1
= 0.1, v
M2
= 0.1, and v
P
= 0.8, and the pool sizes of metabolites
S, M1, M2 and P are set to 1. This plot reveals that the dynamics depend on the pool size in
addition to the flux distribution and provide valuable information on the relative pool sizes that
can be used for dynamic labeling experiments. The pool size does not affect the steady-state
distribution.
15.4 DETAILED EXAMPLE OF THE BNGL/BioNetGen-BASED
APPROACH FOR THE CALVIN CYCLE
The Calvin cycle is a series of biochemical reactions required for carbon fixa-
tion. The carbon source for this cycle is CO
2
. Five metabolites D-ribulose-1,5-
bisphosphate (R5P),
D-erythrose 4-phosphate (E4P), 3-phospho-D-glycerate (G3P),
(2R)-2-hydroxy-3-(phosphonooxy)-propanal (T3P), and
D-fructose 6-phosphate
(F6P) link this pathway with central metabolic pathways. Figure 15.5 provides a net-
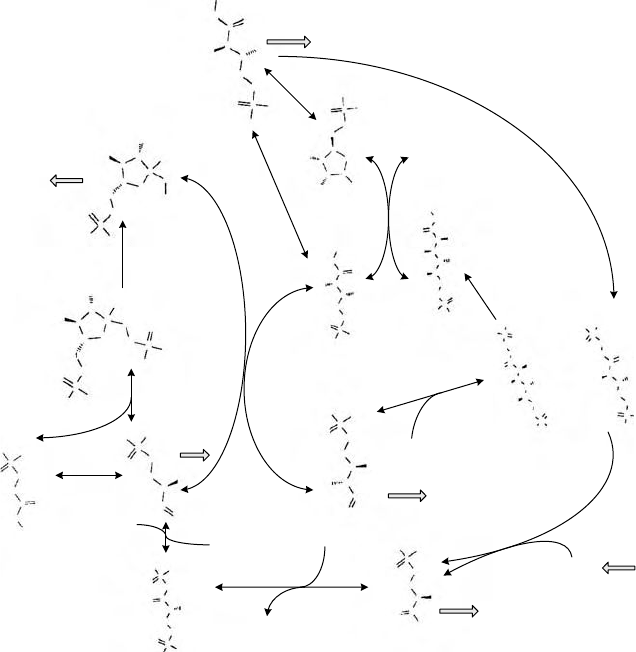
work rendition of the Calvin cycle. The program we developed to visualize chemical
structures, such as that shown in Figure 15.5, uses routines available in the Chemistry
Development Kit [37]. The catalysts involved in the Calvin cycle are functionally
equivalent to those used in other pathways, including the pentose phosphate path-
way and in gluconeogenesis, and therefore, this example may be of interest to a
broad community. The Calvin cycle is of particular interest because this network
involves only a single-carbon input metabolite, and therefore steady-state labeling
Using Systems Biology Techniques to Determine Metabolic Fluxes 415
F6 P
F16P
DHAP
T3P
R5P
R5DP
E4P
G3 P
NAD
NADPH
CO
2
ADP
ATP
R01512
X5P
DHAP
T3P
R01523
R00024
R01063
R01015
R01068
R00762
R01067
R01829
R01845
R01641
R01529
R01056
Ri5P
S7P
S17P
P13G
O
O
O
O
P
P
O
O
3
O
O
O
P
O
2
3
O
O
O
1
O
O
O
O
O
2
4
1
3
O
O
P
O
O
1
1
O
O
O
O
O
O
4
4
4
4
5
3
2
2
2
2
1
1
3
5
7
6
5
5
3
6
7
3
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
P
P
P
O
O
O
O
O
O
O
P
P
P
O
O
O
O
O
2
3
1
O
P
O
O
O
O
O
2
3
1
O
P
O
O
O
O
O
O
O
O
O
O
O
2
3
1
4
5
6
O
P
P
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
O
1
2
3
5
4
O
O
O
O
2
1
3
1
2
3
4
4
5
5
6
O
P
P
P
2
1
O
O
O
O
FIGURE 15.5 The Calvin cycle. The following abbreviations are used for metabolites in
the network: R5P for
D-ribulose 1,5-bisphosphate, G3P for 3-phospho-D-glycerate, P13G for
3-phospho-
D-glyceroyl phosphate, and T3P for (2R)-2-hydroxy-3-(phosphonooxy)-propanal,
DHAP for glycerone phosphate, F16P for
D-fructose 1,6-bisphosphate, F6P for D-fructose
6-phosphate, E4P for
D-erythrose 4-phosphate, X5P for D-xylulose 5-phosphate, S17P for
D-sedoheptulose 1,7-bisphosphate, S7P for D-sedoheptulose 7-phosphate, Ri5P for D-ribose
5-phosphate, and R5P for
D-ribulose 5-phosphate. The KEGG ID is used for reaction identifi-
cation. There is an input flux feeding the pool of CO
2
and five flux sinks from pools of R5P,
E4P, G3P, T3P, and F6P. For each metabolite, the carbon index shown in the figure is based
on InChI™ naming. The indices are used to define the carbon atom fate maps as shown in the
BioNetGen input file.
of CO
2
is completely uninformative. Analysis of the transient labeling patterns of
metabolites is required to learn anything about the internal fluxes from a
13
C-labeling
experiment.We employ the BNGL/BioNetGen-based approach to determine the intra-
cellular fluxes for the Calvin cycle. The BioNetGen input file shown below specifies
a model and simulation for labeling dynamics in the Calvin cycle (Figure 15.5) when

416 Handbook of Chemoinformatics Algorithms
a feed of unlabeled CO
2
is replaced with a mixture of unlabeled and
13
C-labeled
CO
2
. The raw carbon fate maps need to be processed before they can be used as
BioNetGen input. This preprocessing is described in Ref. [19]. The model tracks 562
isotopomers. In the simulation, the system is taken to be in a steady state. At time
t = 0, the composition of the CO
2
feed is changed, such that 10% of incoming CO
2
is labeled with
13
C. The steady-state fluxes in the system remain unchanged (up to
isotope effects, which are expected to be small), but the
13
C labeling pattern of each
intermediate metabolite changes until eventually all intermediates are fully labeled.
BioNetGen is capable of simulating the labeling dynamics. BioNetGen can also gen-
erate a specification of the same model and simulation in M-file and Systems Biology
Markup Language (SBML) formats, which can then be interpreted by other software
tools capable of simulating ODEs, such as MATLAB
®
. For reversible reactions, we
need to specify two fluxes: a forward flux and a backward flux. Instead of using
forward and backward fluxes, reversible reactions can also be modeled using a net
flux and an exchange flux [27]. That is,
v
net
= v
forward
−v
backward
and v
exchange
= min(v
forward
, v
backward
). (15.39)
The exchange flux can be in the range [0, ∞]. The characterization of forward and
backward fluxes as net and exchange fluxes provides a measure of the reversibility
of a reaction, where an exchange flux of 0 corresponds to irreversible reactions and
infinity (∞) indicates fast reaction equilibrium. Isotopomer distributions are not very
sensitive to exchange fluxes. Because it has been shown that the exchange flux can
only be determined to within an order of magnitude [9], an exchange flux can be
transformed into the so-called [0, 1]-exchange flux,
v
exchange
∈[0, 1]=
v
exchange
(1 −v
exchange
)
. (15.40)
In addition, when the exchange flux is very large, numerical instabilities may arise.
In these cases, this range can be specified as, for example, [0.1, 0.9] to remove the
possibility that an exchange flux is either too small or too large. As a measure of the
efficiency of BioNetGen for a problem with some computational complexity (562
isotopomers), we have calculated the CPU time requirements for the Calvin cycle
simulation. On an Intel
®
Pentium
®
4 CPU 3.2 GHz machine, the entire simulation
took a total of 33.8 s. It took 14.4 s to generate the isotopomer balance equations from
the BNGL-encoded input and 17.2 s to equilibrate the system, which corresponds to
the first simulate_ode command. The second simulate_ode command runs another
ODE simulation to identify the isotopomer fractions and took 2.2 s. The computational
cost of simulating a set of stiff differential equations typically scales as N
3
, where
N is the number of equations (number of species). In general, N and the number of
isotopomer balance equations will tend to increase exponentially with the number of
reactions in the metabolic network. However, for a recently reported kinetic Monte
Carlo method [38], the cost of simulation is independent of the number of isotopomers
and scales with the number of reactions (rules).
Using Systems Biology Techniques to Determine Metabolic Fluxes 417
begin parameters
#Pools
V_R5P 1.0
V_R5DP 1.0
V_CO2 1.0
V_G3P 1.0
V_P13G 1.0
V_T3P 1.0
V_DHAP 1.0
V_F16P 1.0
V_F6P 1.0
V_E4P 1.0
V_X5P 1.0
V_Ri5P 1.0
V_S17P 1.0
V_S7P 1.0
# Fluxes
v_R00024 1.00
v_R01512 1.95
v_R01063_f 0.05
v_R01063_b 2.00
v_R01015_f 0.86
v_R01015_b 0.10
v_R01068_f 0.15
v_R01068_b 0.58
v_R00762 0.43
v_R01067_f 0.50
v_R01067_b 0.12
v_R01829_f 0.20
v_R01829_b 0.53
v_R01845 0.33
v_R01641_f 0.43
v_R01641_b 0.10
v_R01529_f 0.10
v_R01529_b 0.82
v_R01056_f 0.50
v_R01056_b 0.17
v_R01523 1.00
vs_T3P 0.05
vs_F6P 0.05
vs_R5P 0.05
vs_E4P 0.05
vs_G3P 0.05
v_vi_CO2_unlabeled 1
v_vi_CO2_labeled 0
end parameters
begin species
R5P(C1 ∼0,C2 ∼0,C3 ∼0,C4 ∼0,C5 ∼0) V_R5P
R5DP(C1 ∼0,C2 ∼0,C3 ∼0,C4 ∼0,C5 ∼0) V_R5DP
CO2(C1 ∼0) V_CO2
G3P(C1 ∼0,C2 ∼0,C3 ∼0) V_G3P
P13G(C1 ∼0,C2 ∼0,C3 ∼0) V_P13G
T3P(C1 ∼0,C2 ∼0,C3 ∼0) V_T3P
DHAP(C1 ∼0,C2 ∼0,C3 ∼0) V_DHAP
F16P(C1 ∼0,C2 ∼0,C3 ∼0,C4 ∼0,C5 ∼0,C6 ∼0) V_F16P
F6P(C1 ∼0,C2 ∼0,C3 ∼0,C4 ∼0,C5 ∼0,C6 ∼0) V_F6P
E4P(C1 ∼0,C2 ∼0,C3 ∼0,C4 ∼0) V_E4P
X5P(C1 ∼0,C2 ∼0,C3 ∼0,C4 ∼0,C5 ∼0)
V_X5P