Dionne G.F. Magnetic Oxides
Подождите немного. Документ загружается.

110 3 Magnetic Exchange in Oxides
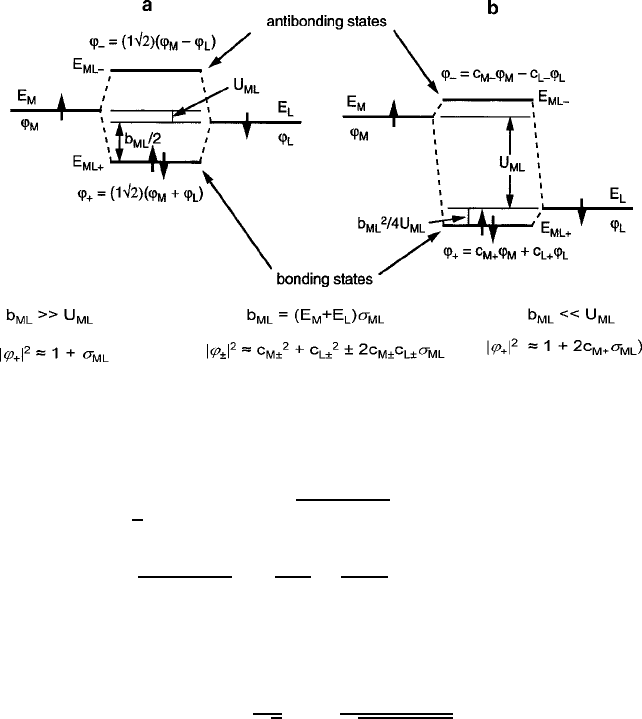
Fig. 3.2 One-electron metal–ligand molecular orbital diagram illustrating the stabilization of an
electron spin pair and the formation of bonding and antibonding hybrid eigenstates for (a) b
ML
>>
U
ML
,and(b) b
ML
<< U
ML
E
˙
1
2
.E
M
C E
L
/ ˙
q
U
2
ML
C b
2
ML
.E
M
C E
L
/
2
˙
U
ML
2
C
b
2
ML
4U
ML
for b
2
ML
=U
2
ML
<< 1; (3.1)
where U
ML
D E
L
–E
M
. Hybrid eigenfunctions are
ML˙
D c
M˙
M
˙ c
L˙
L
,
where
c
M˙
D c
L
1
p
2
0
B
@
1 ˙
U
ML
q
U
2
ML
C b
2
ML
1
C
A
1=2
: (3.2)
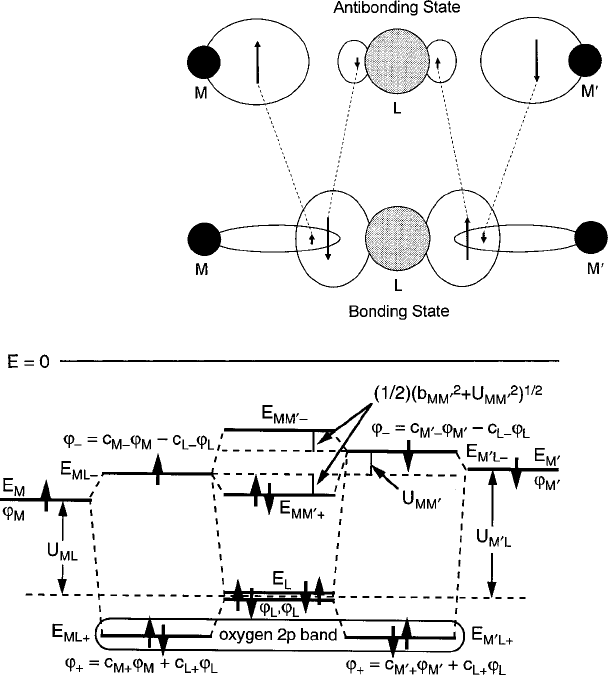
This approximation can be used as a building block to create an artificial M–L–M
0
molecule that represents the actual situations sketched in Fig. 3.3. By attaching a
second cation to the opposite lobe of the same ligand orbital function, we can con-
sider forming an illustrative model by pursuing a qualitative line of reasoning. To
reduce this three-body problem diagrammed in Fig. 3.4 to a manageable two-body
case, an effective overlap integral between M and M
0
must be deduced from the
bonding state of the M–L–M
0
molecule. Once this interaction parameter is deter-
mined, the M–M
0
coupling can be approximated in the manner of a simple diatomic
molecule.
From the MO diagram of Fig. 3.2, the unpaired 3d spins in the e
g
states are
localized to their respective nuclei in the '
ML
and '
M
0
L
antibonding states. The
smaller b
ML
2
=4U
ML
10Dq is, the closer the levels will approach those of a “free”
ion. As long as the b
ML
, b
ML
0
¤ 0, however, a portion of the j'
M
j
2
and j'
M
0
j
2
den-
sities is hybridized with the j'
L
j
2
density in the lower energy bonding state. As a
3.1 Interionic Magnetic Exchange 111
Fig. 3.3 One-dimensional
sketches of
metal–oxygen–metal
molecular orbital hybrid
states illustrating the
fractional spin distributions of
the respective 3d metal and
2p ligand wavefunctions
Fig. 3.4 One-electron metal–oxygen–metal molecular orbital diagram illustrating the formation
of the basic bonding and antibonding hybrid eigenstates that produce superexchange in magnetic
oxides. Figure reprinted from G.F. Dionne, J. Appl. Phys. 99, 08M913 (2006) with permission.
c
2006 by the American Institute of Physics
result, the two 3d spins are partially delocalized into antiferromagnetic states by an
effective overlap integral
MLM
0
that is mediated through the ligand 2p orbital func-
tion containing its original paired spins. As depicted schematically in Figs. 3.1d, e,
excess densities in the regions between the cations and anion, i.e., (1 C
ML
), will
define the volume of bonding interaction, while depleted densities (1–
ML
/ in the
corresponding antibonding states will restore the population total (see Appendix 3A
of Chap. 2). From this diagram, it is evident that the orbital stabilization occurs in
the bonding state, despite the fact that the 3d spins are mainly localized in the anti-
bonding state. The effective overlap integral
MLM
0
can then be approximated from
the volumes of the two 3d wavefunctions represented in the bonding states j
MLC
j
2
and j
M
0
LC
j
2
, i.e.,
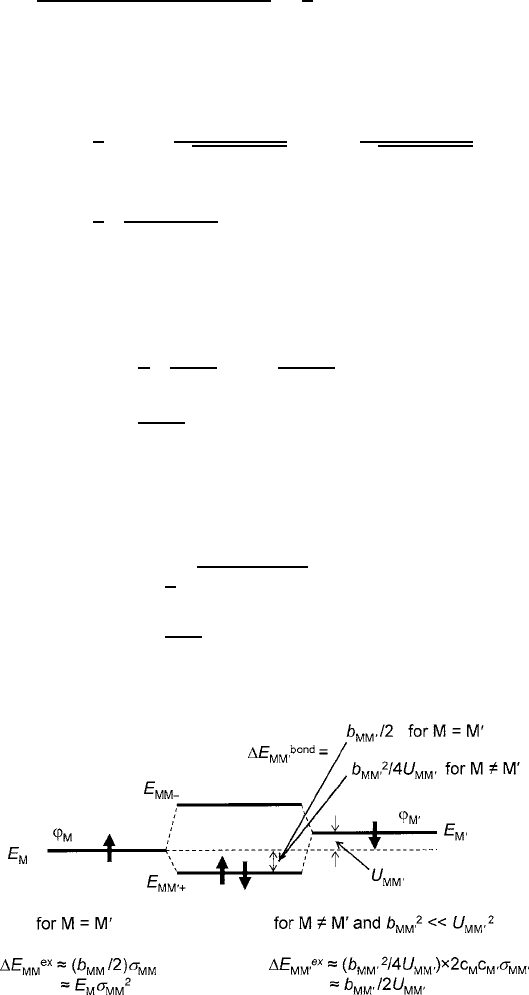
112 3 Magnetic Exchange in Oxides
MLM
0
c
MC
c
LC
ML
C c
M
0
C
c
LC
M
0
L
c
2
MLC
C c
2
M
0
LC
1
2
.c
MC
c
LC
ML
C c
M
0
C
c
LC
M
0
L
/;
(3.3)
where
c
MC
c
LC
1
2
2
6
4
0
B
@
1
U
ML
q
U
2
ML
C b
2
ML
1
C
A
0
B
@
1 C
U
ML
q
U
2
ML
C b
2
ML
1
C
A
3
7
5
1=2
D
1
2
b
2
ML
U
2
ML
C b
2
ML
1=2
: (3.4)
For a magnetic oxide with strong ionic bonding, b
2
ML
=U
2
ML
<< 1 and c
MC
c
LC
b
ML
=2U
ML
,sothat
MLM
0
1
2
b
ML
2U
ML
ML
C
b
M
0
L
0
2U
M
0
L
0
M
0
L
0
b
ML
2U
ML
ML
for homonuclear M D M
0
: (3.5)
Thus, the effective overlap integral between M and M
0
can now be labeled as
MM
0
D
MLM
0
, and the simplified two-body problem depicted in Fig. 3.5 can be
solved with
˙
D c
M˙
M
˙ c
M
0
˙
M
0
and a bonding stabilization energy
E
bond
MM
0
1
2
q
U
2
MM
0
C b
2
MM
0
b
MM
2
for homonuclear M D M
0
; (3.6)
Fig. 3.5 Two-body approximation to the metal–oxygen–metal molecular orbital extracted from
the upper half of Fig. 3.4

3.1 Interionic Magnetic Exchange 113
where b
MM
0
D .E
M
C E
M
0
/
MM
0
. Because only the delocalized fraction of the
3d spin population in
h
'
C
j
'
C
i
is required to satisfy Pauli spin pairing, the por-
tion of the covalent-bonding energy that stabilizes the antiparallel spin alignment is
weighted by the delocalized fraction 2c
MC
c
M
0
C
MM
0
.From(3.4)and(3.6)
E
ex
MM
0
D E
bond
MM
0
2c
MC
c
M
0
C
MM
0
D
1
2
q
U
2
MM
0
C b
2
MM
0
b
2
MM
0
U
2
MM
0
C b
2
MM
0
!
1=2
MM
0
D
b
MM
0
2
MM
0
; (3.7)
which is a first-order function of b
MM
0
.
In terms of the Heisenberg exchange energy between ions M and M
0
for com-
bined electron spins E
ex
MM
0
D2J
MM
0
S
M
S
M
0
,(3.7) for a single orbital spin pair
where S
M
D 1=2 can be expressed as J
MM
0
D b
MM
0
MM
0
. Because the screening
parameter h
MM0
is presumed to be positive based on the ground state spin occupa-
tion in Fig. 3.5, antiferromagnetism should prevail and J
MM
0
would have a negative
sign in the Heisenberg model. However, in situations where direct overlap between
other M and M
0
orbitals can compete, h
MM
0
could be negative, causing an inversion
of the bonding/antibonding states and a weakening or even reversal in sign of J
MM
0
leading to an antibonding ferromagnetic ground state.
To ensure the ferromagnetic alignment where b
MLM
0
is small, the missing con-
tribution from the e
2
=r
ij
repulsion would have to be introduced by a two-electron
exchange calculation, which would make a positive addition to the transfer element
and lead to a reversal in the cation–cation bonding/antibonding states '
M
and '
.
It is important to recognize that although these parameters are not the same as those
used in the more formal analysis of superexchange, at a qualitative level the model
provides helpful insight as well as providing an estimate of the effective transfer
integral b
MM
0
. It also allows the addition of competing direct cation–cation b inte-
grals to arrive at a combined result. A comparison of the two approaches is offered
in the next section. Recall also that since b
ML
varies directly as the first power of the
overlap integral
ML
, by definition in (2.38), both
MM
0
and the quasi-direct trans-
fer integral b
MM
0
are proportional to
ML
2
as indicated by (3.5). When the actual
exchange stabilization energy E
ex
MM
0
in (3.7) is considered, its dependence on the
cation–anion orbital overlap becomes
ML
4
, as pointed out also by Ballhausen [4].
This high sensitivity of the indirect exchange coupling confirms that the stability
of long-range spin ordering in a crystal lattice is severely diminished by magnetic
dilution.
3.1.2 Valence-Bond Solutions
Analysis of interionic electronic and magnetic properties in ionic crystal lattices
by the molecular-orbital (MO) approach is largely conceptual, based on probabi-
lity estimates of the ground-state electron spin occupancy. As the name implies, a

114 3 Magnetic Exchange in Oxides
molecule is first built from the strong Coulomb electric fields between cations and
anions, and then as demonstrated in the previous section, a second cation is added
to estimate the strength of magnetic exchange coupling that is more than an or-
der of magnitude weaker than the electrostatic bonding. Because the stabilization
from electronic hybridization is controlled by the wavefunctions overlap volumes,
the orbital overlap integral represents a measure of the delocalization and is re-
tained explicitly in the formalism. In its purest form, however, the method does not
readily extend to collective electron situations unless the presumption of long-range
homogeneity allows the adoption of Hartree-Fock formalisms that lead to band mod-
els. An important feature of the MO approach is the preservation of the internal
electronic energy-level information of local cations from which the sensitivity of
the orbital angular momentum to lower symmetry crystal fields affects the multi-
plet structure that in turn determines the local magnetoelastic, magneto-optical, and
magnetic relaxation properties.
For the discussion of specific molecules, particularly as their complexities in-
crease, the more rigorous valence-bond (VB) approach offers a more quantitative
methodology [5]. Computation of bonding and exchange energies for specific
structures can be carried out by a method that allows the introduction of multi-
electron wavefunctions and includes the mutual electron repulsive energy e
2
=r
ij
.
A two-electron Hamiltonian that retains the nuclear and electronic interactions
is a convenient starting point. The effects of crystal fields on the partially filled
d -electron states can also be included if the appropriate molecular orbitals are em-
ployed as the starting wavefunctions. For the analysis of direct magnetic exchange
(applied originally to the H
2
molecule [1]), the valence-bondmethod is used to com-
pute the orbital interaction energies that dictate the high-spin (parallel) and low-spin
(antiparallel) states based on the indistinguishability requirement (Pauli principle)
of the two electrons sharing hybrid orbital states of both atoms. For this purpose,
the Coulomb energy terms E
M
and E
L
of (3.1) are replaced by the actual interionic
electron–nuclear attraction and electron–electron mutual replusion terms similar to
obtain a two-electron Hamiltonian H D H
0
C H
1
,where
H
0
D
„
2
2m
e
r
2
a
Cr
2
b
Z
a
e
2
r
a1
Z
b
e
2
r
b2
C
Z
a
Z
b
e
2
r
ab
;
H
1
D
Z
a
e
2
r
a2
Z
b
e
2
r
b1
C
e
2
r
12
: (3.8)
In the MO solutions, such as the hydrogen molecule ion sketched in Fig. 3.6a, H
1
contains only the nuclear terms. Under this restriction, the results for the MO and
VB methods are usually identical. However, that quickly changes when the electron
correlation energy is introduced in a formal manner. As defined in Fig. 3.6b, the
distances r
a2
and r
b1
are the electron–nuclear separation of the exchanged electrons
labeled 1 and 2 on nuclei a and b, respectively, and r
12
is the separation between the
electrons themselves. Two-electron degenerate eigenfunctions '
0
D '
a1
'
b2
(initial
occupancy) and '
ex
D '
a2
'
b1
(exchanged occupancy) of this Hamiltonian are con-
structed from products of single-electron functions for computation of the matrix
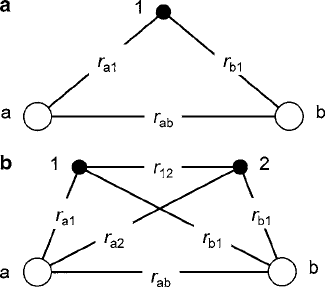
3.1 Interionic Magnetic Exchange 115
Fig. 3.6 Electrostatic
linkages between nuclei a and
bandelectrons1an2for(a)
the single electron hydrogen
molecule ion H
C
2
,and(b)the
hydrogen molecule H
2
elements K
12
.Coulomb/ Dh'
0
jH
1
j'
0
i and J
12
.exchange/ Dh'
0
jH
1
j'
ex
i of the
2 2 Slater determinant described in Sect. 1.3. The solutions of the secular equation
give bonding and antibonding states energies according to K
12
˙J
12
. Consequently,
the sign of J
12
determines whether the spins in the ground state are parallel .S D 1/
for J
12
>0, or antiparallel .S D 0/ for J
12
<0.
The computation of J for a superexchange coupling between two magnetic ions
(spin cations) M
nC
a
and M
nC
b
bonded to a common O
2
ligand begins with the
assumption that unfilled orbital states are present in the M
a
–O–M
b
molecule. Since
the electrostatic stabilization among ions is maximized if the oxygen ligands carry
2– charges, meaning that the oxygen 2p band is filled, the unpaired electron spins
reside on the metal cations. Because of the ionic character of the bonding and the
important influence of crystal fields that remove the degeneracies of the 3d shell
states, as described in Chap. 2, the analysis of the interactions between M
a
and M
b
are treated on an individual orbital state basis.
A physical rationale is proposed to serve as a basis for superexchange second-
order perturbation analysis, which requires the existence of an excited state that
can be mixed with a state of lower energy by the electrostatic perturbation of
the molecular bonding, i.e., a nonzero off-diagonal matrix element created by a
transfer integral, to produce separate bonding and antibonding states. Maintain-
ing observance of the Pauli principle, the model sketched in Fig. 3.7ashowsthe
transfer of spins from the O
2
2p states onto the unfilled orbitals of the cations
to produce stabilization of an antiferromagnetic state between the spins localized
on the two cations through the creation of a virtual excited state energy U .Ini-
tially both spins are resident on the respective cations, with the relevant 2p orbital
state of the O
2
ion filled. The excited state is postulated by creating a virtual
transfer of the M
a
spin to the equivalent M
b
orbital state, resulting in the reaction
M
a
nC
C M
b
nC
! M
a
.nC1/C
C M
b
.n1/C
C U. A return reaction in the will com-
plete the transfer of spins from '
0
D '
a1
b2
(initial occupancy) to '
ex
D '
a2
'
b1
(exchanged occupancy), and stabilize antiferromagnetic spin alignment.
If the starting wavefunctions are molecular orbitals that reflect the effects of the
cross electron–nuclear terms of (3.8), the electrostatic perturbation H
1
can now be
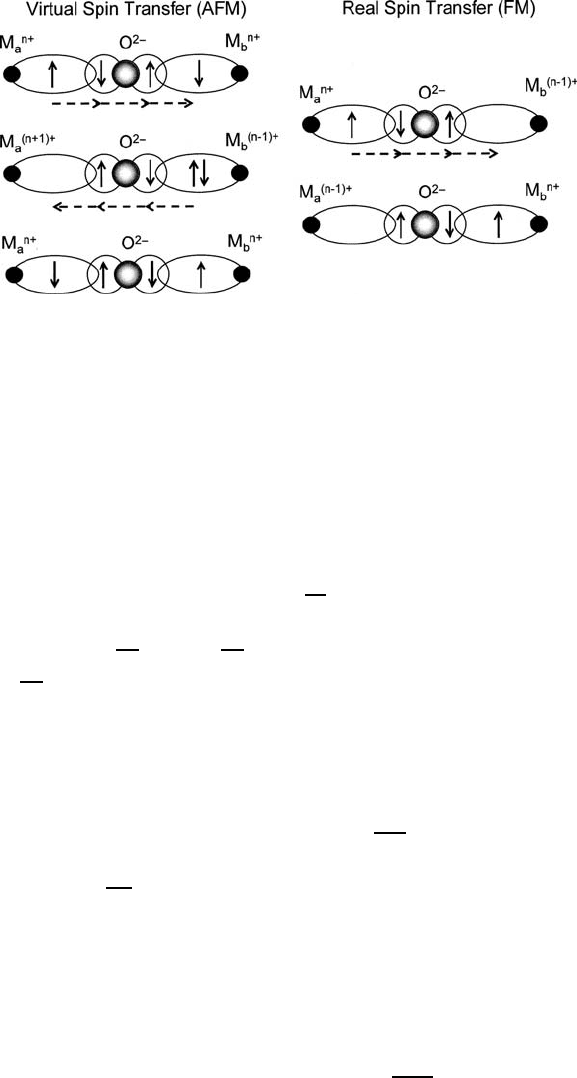
116 3 Magnetic Exchange in Oxides
Fig. 3.7 Tutorial illustration of superexchange virtual and real spin transfer: The former produces
antiferromagnetism, the latter, somewhat weaker ferromagnetism accompanied by electrical con-
ductivity
approximated by the e
2
=r
12
electron repulsion term. This approach was used by
Anderson in his original formalization of the superexchange interaction [6]. For
apairof3d orbital states of cations linked by a 2p orbital of an oxygen ligand,
second-order perturbation theory is applied to the system to compute the energy
of the stabilized ground state. The standard correction to the orbital ground-state
energy for the orbital energy is then
E D
b
2
U
; (3.9)
where b
2
D
h
1
j
e
2
r
12
j
2
ih
2
j
e
2
r
12
j
1
i
, U D
h
0
j
V
12
j
ex
ih
ex
j
V
12
j
0
i
,and
V
12
D
e
2
r
12
exp.r
12
/, with as a screening parameter [7]. After the addition of
the electron spin exchange operator s
1
s
2
of negative sign (for antiparallel align-
ment) from Anderson’s formal solution by second quantization theory and the use
of the four Fermi spin operators to account for the two anion spins occupying the
2p orbital, (3.9) becomes
E
virtual
ab
D2J
virtual
ab
s
1
s
2
D 2
2b
2
U
s
1
s
2
; (3.10)
where J
virtual
ab
D
2b
2
U
. This result has the reverse sign from the first-order result for
direct exchange
E
direct
ab
D2J
direct
ab
s
1
s
2
; (3.11)
where J
direct
ab
D
h
'
0
j
H
1
j
'
ex
i
>0.
In generalized format for n orbital levels linking two cations i and j,theex-
change stabilization energy derived from the spin operator part of (3.9)is
E
virtual
ij
D2J
virtual
ij
S
i
S
j
D 2
X
n
2b
n2
ij
U
n
s
i
s
j
; (3.12)
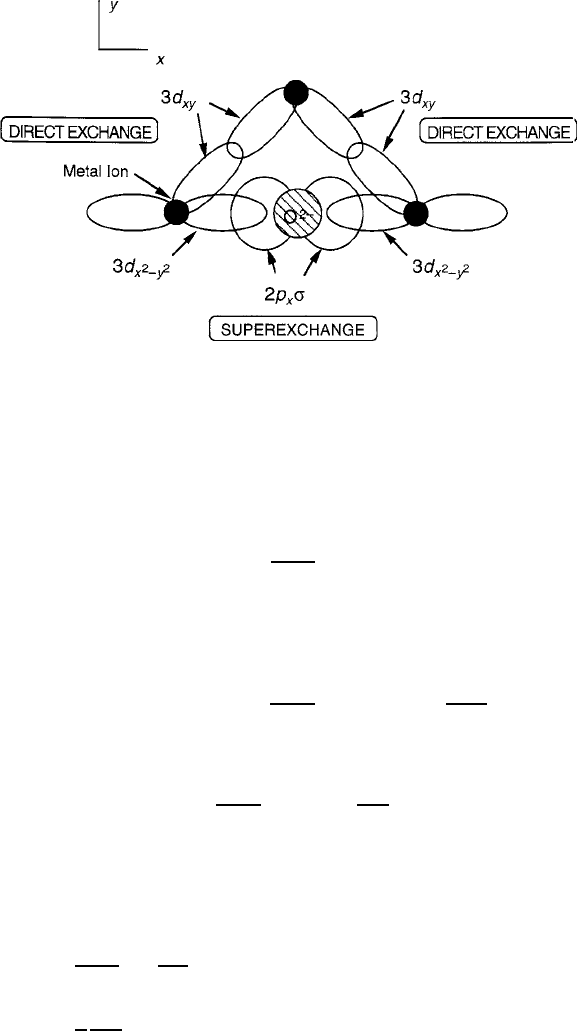
3.1 Interionic Magnetic Exchange 117
Fig. 3.8 Metal–oxygen exchange bonding showing the comparison between direct and indirect
or superexchange. Superexchange stabilizes antiferromagnetism; direct exchange is commonly
ferromagnetic
where J
ij
is the direct exchange contribution suggested by Fig.3.8. If ions have total
spins S
i
and S
j
summed over n orbital states with s
i
D s
j
D 1=2, we can write
s
i
s
j
D
1
4S
i
S
j
S
i
S
j
; (3.13)
and the combined exchange constant for the molecule is determined by applying
(3.11)and(3.12):
J
ij
D J
direct
ij
C J
virtual
ij
D
1
4S
i
S
j
X
n
J
ndirect
ij
2b
n2
ij
U
n
ij
!
;
or simply
J
ij
D
1
4S
i
S
j
X
n
J
n
2b
2
n
U
n
; (3.14)
where the subscript in (3.12) have been replaced by n for the parameters J and b.
For superexchange, we can extract from (3.12)forS
i
D S
j
D S,
or
J
virtual
ij
D
1
2S
i
S
j
X
n
b
n2
ij
U
n
J
virtual
n
1
n
2b
2
n
U
n
for n equivalent orbital states with equal S D n=2; (3.15)

118 3 Magnetic Exchange in Oxides
Table 3.1 Bonding and exchange parameters
Molecular orbital(MO) Valence bond(VB)
ML
a
0.3
b
ML
b
.
E
M
CE
L
/
ML
10 eV b
2
n
D
'
1
ˇ
ˇ
ˇ
e
2
r
12
ˇ
ˇ
ˇ
'
2
ED
'
2
ˇ
ˇ
ˇ
e
2
r
12
ˇ
ˇ
ˇ
'
1
E
1:5 eV
U
ML
E
L
–E
M
15 eV U
c
n
h'
0
jV
12
j'
ex
i15 eV
c
10Dq
ˇ
ˇ
b
2
ML
=4U
ML
ˇ
ˇ
D 1:7 eV
MM
.
b
ML
=2U
ML
/
ML
0:1
b
MM
2E
M
MM
ML
2 eV
E
Super
ex
n
.
1=2
/
b
MM
¢
MM
0:1 eV
.
n D 1
/
E
Super
ex
nb
2
n
=U
n
0:1 eV
.
n D 1
/
J
Super
ex
nb
MM
MM
D0:19 eV
.
n D 1
/
J
Super
ex
.
1=n
/
2b
2
n
=U
n
D0:2 eV
.
n D 1
/
a
Per e
g
orbital in an octahedral site, 180
ı
bond angles
b
E
M
D10eV , E
L
D25 eV
c
These values are on the order of ionization potentials and electron affinities. See Ref. [7]
and we can also write
E
virtual
ex
X
n
b
2
n
U
n
n
b
2
n
U
n
: (3.16)
The details of the quantum mechanical solution that include the explicit representa-
tion of the Fermi spin operators that control the alignment of the respective electron
spins s
1
and s
2
vectors are discussed in many standard texts [7, 11] and will not be
repeated here. However, it is important to compare the initial assumptions of the
MO and VB models and how they influence the results relevant to crystal field and
magnetic exchange energies (Table 3.1).
2
In typical situations, the spin directions are not precisely aligned, but form an
angle
ij
with each other. To introduce this angular dependence into Anderson’s
result, the use of the exact relation for the scalar product S
i
S
j
is not convenient
because of the quantum correction S.S C 1/ in the angular momentum addition.
If the Ising approximation is adopted, whereby only the z components of S are
retained in the vector product, S
z
D S cos
ij
=2
and the exchange energy for two
spins with z
ij
equivalent neighbors can be expressed as
E
ferro
ex
.VB/ D2z
ij
J
ij
S
i
S
j
D2z
ij
J
ij
S
zi
S
zj
cos
2
ij
2
Dz
ij
J
ij
S
zi
S
zj
1 C cos
ij
; (3.17)
2
There is a distinction between U
MM
0
in the MO case and U of the VB method. In the MO case,
U
MM
0
represents the in-lattice difference in energy between the two outermost electrons that remain
with their parent ions, and exchange is viewed simply as a small first-order perturbation without
allowing for the formation of new ionic states by formal charge transfer. In the VB case, an exci-
tation defines U as the net change of two ionic stabilization energies involved in the virtual spin
transfer, approximated by the difference in the ionization potentials between the initial and final
valence states of the ions. Here the perturbation is treated mathematically as second order.Asa
result, U
MM
0
0 in the localized MO treatment for like cations M D M
0
, but for the virtual spin
transfer of the VB method, U ¤ 0.SeeTable 3.1.

3.1 Interionic Magnetic Exchange 119
and for the antiparallel case,
E
antiferro
ex
.VB/ D2z
ij
J
ij
S
i
S
j
2z
ij
J
ij
S
zi
S
zj
sin
2
ij
2
(3.18)
D z
ij
J
ij
S
zi
S
zj
1 cos
ij
:
To provide stabilization for ferromagnetism (
ij
D 0), J
ij
>0and for antiferro-
magnetism
ij
D
, J
ij
<0. From this general result, the various spin ordering
situations that occur in transition-metal oxides can be examined.
This result was also obtained by Goodenough [7], who reasoned that Hubbard’s
exchange integral b
ij
[12] could be modified to obey the Pauli exclusion principle by
constructing a transfer integral t
12
for a particular pair of orbital states that included
the canting angle
ij
between ionic spins. For this exercise, the relations are t
ij
D
b
ij
cos
ij
=2
and b
ij
sin
ij
=2
for antiparallel and parallel alignment, respectively.
Installation of the t
12
expressions in place of b
ij
in the superexchange terms of (3.12)
will yield results equivalent to (3.17)and(3.18).
If the MO model is used, the transfer integral is a first-order perturbation and the
respective relations of (3.17)and(3.18) simplify to
E
ex
.MO/ D2z
ij
J
ij
S
zi
S
zj
cos
ij
; (3.19)
where parallel alignment (
ij
D 0) requires J
ij
>0, and antiparallel alignment
ij
D
sets J
ij
<0. This approach can be simply the analysis when first-order
double exchange interactions are also present.
3.1.3 Spin Alignment in Oxides
The foregoing analyses for estimating spin alignment can be summarized. Ferro-
magnetism (parallel spins) occurs when the collective electron repulsion energy
P
e
2
=r
ij
is dominant in H
1
of (3.8). This is expected when the nuclear separa-
tion r
ab
is large enough that the electron orbits overlap enough to disperse the spins
according to Hund’s rule without being unduly influenced by the attraction fields of
their opposite nucleus (antibonding state is lowest). To comply with (3.12), this con-
dition requires that J>0. It should be noted in Fig. 1.12 that the light members of
the 3d series provide fewer numbers of electrons to screen the attractive effects of
the nuclei for ferromagnetism to occur. This observation is also consistent with the
shorter bond lengths found for Ti, V, Cr, and Mn. Antiferromagnetism (opposing
spins) occurs when the interionic electron–nuclear terms of (3.8) are dominant in
H
1
. This is expected when the nuclear separation is small enough that the electron
charge clouds overlap greatly .r
ab
<< r
a2
;r
b1
/ and the dispositions of the spins are
controlled by the attractive fields of the opposite nuclei (bonding state is lowest). For