Seminario J.M. Molecular and Nano Electronics. Analysis, Design and Simulation
Подождите немного. Документ загружается.

64 Ying Luo et al.
Ground-state
Excited-state
f
E
f
G
shift
(A)
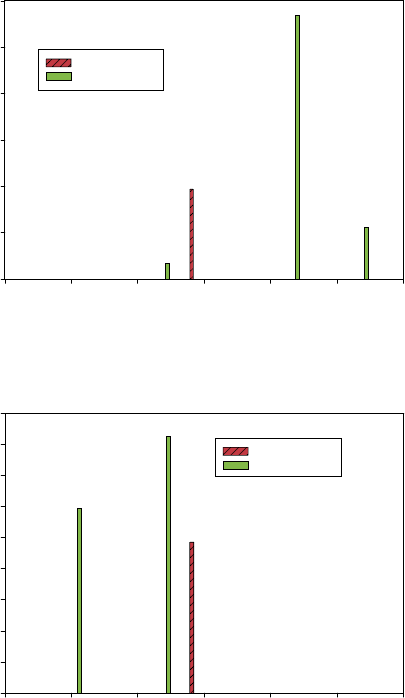
(B)
Phonon Absorption Spectrum
0
10
20
30
40
cis -2-Butene
trans-2-Butene
Normalized Intensity
cis - Same side of the double bond
trans - Opposite sides of the double bond
Wavenumber (cm
–1
)
0 500 1000 1500 2000
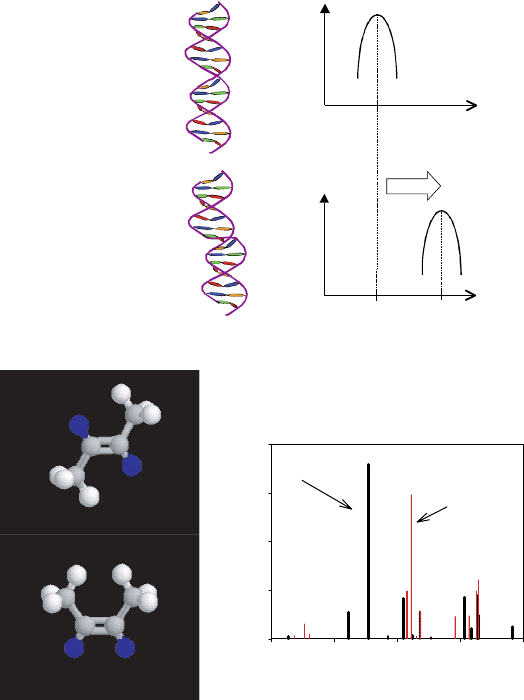
Figure 5 Basic ETO effect: (A) hypothetical DNA example, (B) isomers of butane and simulated
spectra [1]
2.2.1. Simulation and analysis of butene isomers
In the initial study, the cis-trans isomers of butene have been chosen because they
are very well known and simple examples of a ground- and metastable-state pair with
significantly different geometries. The cis-trans isomers are geometric isomers, a type
of stereoisomerism in which atoms or groups display orientation differences around a
double bond, such as trans-2- and cis-2-butene as was shown earlier in Figure 5(B). This
work demonstrated molecular isomers that yield different sets of vibrational frequencies.
In this case to be studied, photo-induced transitions bring about the conversion from
one geometric shape (i.e., trans-2-butene) to another (i.e., cis-2-butene) through rotation
about a double bond and the conversion is also called isomerization.
The analysis presented here includes calculations of the optimized energies, vibra-
tional frequencies, and infrared intensities that were carried out at the Hartree–Fock
Bio-molecular devices for terahertz frequency sensing 65
(HF) level within the split valence polarized 6-31G(d) basis set in the Gaussian 98 pack-
age [31]. The energies of the excited states were found within the single configuration
interaction approach (CIS), where one models the first excited state as combinations of
a single substitution from the highest occupied molecular orbital (HOMO) to the lowest
unoccupied molecular orbital (LUMO). Vibrational frequencies were then computed by
determining the second derivatives of the potential energy with respect to the Cartesian
nuclear coordinates and then transforming to mass-weighted coordinates. Due to the fact
that electron correlation is neglected, the frequencies computed using the HF approxi-
mation are known to be overestimated by approximately 10–12%. Furthermore, because
a medium-sized basis set was used, the derived values can be expected to deviate even
more from experiments, i.e., by approximately 15% in total [32]. Therefore, a scaling
was performed on the originally calculated frequencies by an empirical factor of 0.893
to eliminate known systematic errors in the physical model.
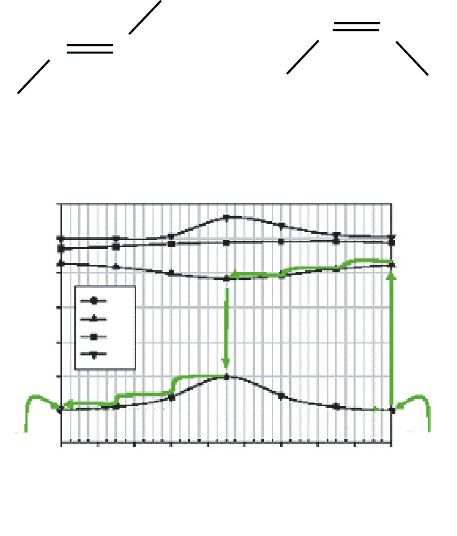
To monitor the process of isomerization from trans-2-butene to cis-2-butene, the
torsional angle , defined as the dihedral angle of the plane of C1–C2–C3 and the
plane of C2–C3–C4 around the double bond in the C1–C2
==
C3–C4 chain associated
with the molecule given in Figure 6(A), was taken to be the reaction coordinate. The
potential energy (PE) curves associated with the isomerization process for the ground
(S
0
and the first excited singlet state (S
1
) of 2-butene are shown in Figure 6(B), along
(B)
2
3
4
1
HC
HC
2
CH
3
CH
3
CH
3
CH
4
H
3
C
H
3
C
1
(A)
θ = 180°, trans-2-butene θ = 0°, cis-2-butene
0
0
2
4
6
8
10
12
20 40 60 80 100 120
S
0
S
2
S
3
S
1
140 160 180
Torsional angle (degree)
Energy (eV)
cis- trans-
Figure 6 (A) Geometrical structure of trans-2-butene and cis-2-butene; (B) Potential energy
curves associated with the isomerization process which involve the ground-state, S
0
, and the first
excited-state, S
1
. Arrows depict the energy-path of the molecules
66 Ying Luo et al.
with the next two excited states S
2
and S
3
. Here it is important to note that the trans-2
geometry (i.e., = 180
) is the natural-state and the cis-geometry (i.e., = 0
)isthe
metastable-state. Without external excitations, the large PE barrier 1.98 eV in the ground
state prevents the transition from trans-2-butene to cis-2-butene. It is also clear from
Figure 6(B) that there is no barrier in the first excited singlet state S
1
, which is ideal for
an ultra-fast switching between the trans- and cis- isomers.
Potential energy curves can be used to illustrate this optically induced isomerization
process by following the superimposed arrows. The process begins with a required
optical excitation (∼846 eV, 146 nm) of the “electronic-state” of the trans-geometry
from S
0
to S
1
. This is followed by a non-radiative decay to the S
1
PE valley minimum,
which corresponds to a 90
rotation about the reaction coordinate. At this point, the
molecule undergoes an “electronic” radiative decay from S
1
to S
0
. This is followed by
a second non-radiative decay to the cis-geometry, which corresponds to a second 90
rotation about the reactive coordinate. At this point, butene will remain in the metastable
cis-2 geometry until thermal relaxation of the system back to the ground state.
To estimate the probability that the excited trans-2-butene will follow the isomer-
ization process described above, instead of relaxing back to its own ground state, we
investigate the molecular dynamics of butene in the first singlet excited state S
1
by
combining the theory of Newton’s Dynamics and standard quantum chemistry software
Gaussian 98. We began the investigation from cis-geometry ( =0
and calculated the
time for the excited cis-geometry to achieve the energy minimum ( = 90
and the
excited trans-geometry ( =180
of S
1
. The calculation procedure is as follows: (i) we
calculated the potential energy of the ground state and the excitation energy of S
1
of the
optimized cis-butene using Gaussian 98 package and by adding the two energies we got
the potential energy of the excited cis-butene. Note that we took this potential energy as
the total energy of butene ( i.e. we assume the kinetic energy of excited cis-butene is 0).
To initiate the rotation, we actually started from a very small with a very small initial
velocity. (ii) For every 10
, we calculated 20 points (i.e., we performed Gaussian 98
calculation every 05
). For every point, we calculated the ground-state energy without
optimization and the corresponding excitation energy; thus we got the potential energy
of the excited state. By subtracting the potential energy from the total energy, we got
the kinetic energy. Then we used the energies of 20 points to calculate the time for
butene to rotate 10
by integrating Newton’s equations.
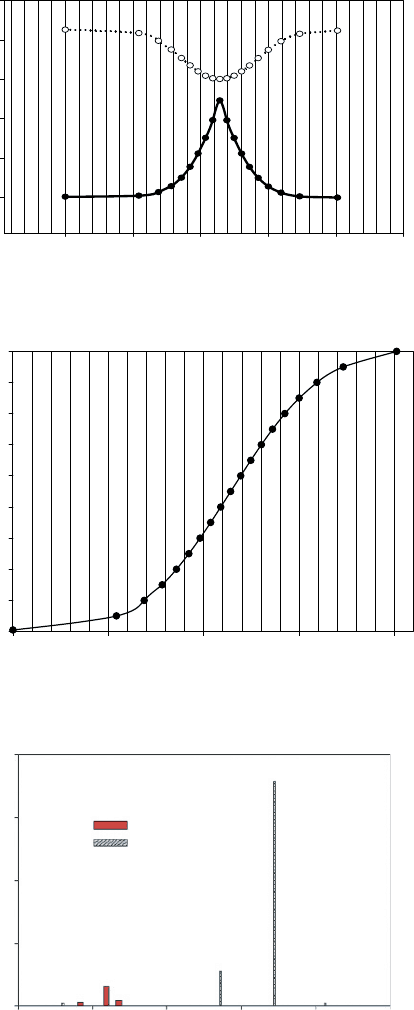
The relation between the potential energies of excited states and time is given in
Figure 7, and for demonstrative purposes, the PE curves of the ground state S
0
is
presented on the same graph. We can see that the time for excited trans-2-butene to
relax to the valley minimum is about 100 fs and symmetrically it takes almost the same
time to go from the minimum to cis-2-butene. On the other hand, according to the
spontaneous emission theory the time for excited trans-2-butene to relax back to ground
state is about 59 ×10
3
s. So the isomerization process will be significantly probable.
In Figure 8, we demonstrate how the dihedral angle of C1–C2
==
C3–C4 double bond, or
the geometric structure of butene, will change with time.
The lowest vibrational frequencies and the associated IR intensities that were cal-
culated for trans-2-butene and cis-2-butene are compared in Figure 9. These results
indicate a significant difference in spectral signatures of the two molecular conforma-
tions and one that has the general quantitative characteristics needed by the previously
discussed bio-molecular architecture.
Bio-molecular devices for terahertz frequency sensing 67
Time (fs)
Energy (eV)
0
2
4
6
8
10
0 50 100 150 200 250
Figure 7 Molecular dynamics of butene: Energies of first singlet excited state vs. time
Time (fs)
Dihedral Angle θ (Degree)
0
20
40
60
80
100
120
140
160
180
0 50 100 150 200
Figure 8 Molecular dynamics of butene: Dihedral angle vs time
40
30
20
10
0
0 200 400 600 800 1000
Vibrational frequencies (cm
–1
)
IR intensities (km /mole)
trans-2-butene
cis-2-butene
Figure 9 The calculated characteristics of the lowest spectral signatures of trans-2-butene and
cis-2-butene [1]

68 Ying Luo et al.
Table 1 A comparison of calculated spectral signatures with measurements of McKean et al. [33]
cis-2-butene trans-2-butene
McKean el al. [33] Calculated in this
work
McKean et al. [33] Calculated in this
work
566 5442 239 2360
686 6878 284 2712
865 9342 9643 9568
970.9 9973 9788 9907
1009.1 10516 10468 10254
1036.9 11296 10636 10529
1268.5 13663 1303 13006
1383.8 14051 13822 13989
1408.4 14173 14474 14519
1444.5 14581 1461 14669
1456.2 14602 28356 28489
1458.9 14669 2871 28900
1669 17029 2890 28901
2893.2 28529 29325 29176
2900.8 28558 29494 29573
2914.5 28919 2973 29619
2929.5 29325
2947.5 29514
2954.1 29556
2979.2 29798
Table 1 compares the calculated frequencies to prior experimental measurements of
McKean et al. [33]. One can see that all the absorption frequencies calculated in this
work are reasonably-good agreeable (i.e., within ∼3%) with the experimental results.
While this study yielded qualitative bio-molecular function of the type needed for
the proposed architecture, the required excitation from S
0
to S
1
is far too large (i.e.,
∼85eV) to be practical, and the associated spectral bio-signatures are well above the
THz regime where species-specific information is expected to be present. Hence, the
alternative molecule retinal will now be considered, which is known to be visible-
frequency light sensitive, and which is more complex and can be expected to yield much
lower frequency vibrational modes.
All-trans-retinal is the chromophore of bacteriorhodopsin, the light transducing pro-
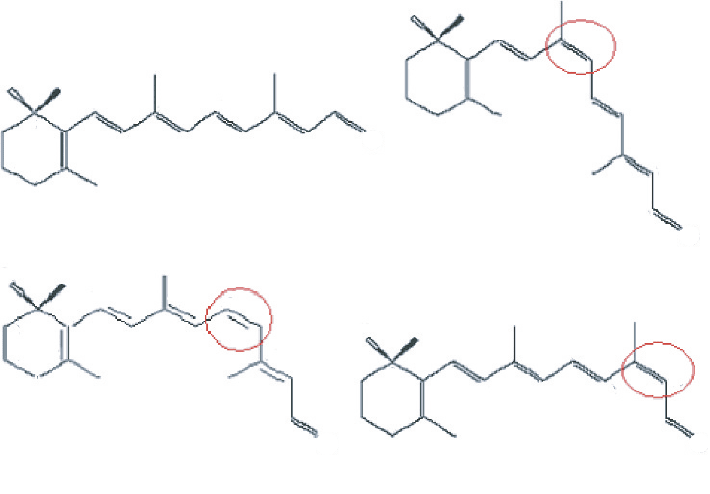
tein in the purple membrane of Halobacterium salinarium. The isomerization from
all-trans to 13-cis conformation takes places very quickly in approximately 500 fs [34]
induced by absorption of a 568 nm photon [35]. 11-cis-retinal is the chromophore of
rhodopsin. The ultra-fast (in ca. 200 fs [34]) photoisomerization from 11-cis-retinal to
all-trans-retinal conformation has been elucidated as induced by absorption of a 498 nm
photon [35]. Besides 11-cis-retinal and 13-cis-retinal, 9-cis-retinal is also a geomet-
ric isomer of all-trans-retinal and derives from the rotation of all-trans-retinal around
C9
==
C10 double bond by 180
although 11-cis-retinal and 13-cis-retinal derive from
the rotation of all-trans-retinal around C11
==
C12 and C13
==
C14 double bonds.
Bio-molecular devices for terahertz frequency sensing 69
16
16
16
17
17
17
7
7
7
7
6
6
1
1
2
2
3
3
4
4
5
5
6
1
2
3
4
5
6
1
2
3
4
5
8
8
8
8
9
9
9
9
10
10
10
10
11
11
11
11
12
12
12
12
13
O
O
OO
13
13
13
14
14
14
14
15
15
15
15
(A) all-trans-retinal (B) 9-cis-retinal
(D) 13-cis-retinal(C) 11-cis-retinal
Figure 10 Geometric structures of retinal isomers
The geometric structures of all-trans-retinal and its isomers are shown in Figure 10.
The retinal has two properties which make it very interesting for our studies. First,
the electrons in retinal readily absorb photons in the visible range of wavelengths
(400–700 nm) which means the molecule can be optically induced into excited-states.
Second, as this study will show, the lowest vibrational frequencies are in THz region.
2.2.2. Simulation and analysis of retinal isomers
The vibrational spectra were calculated at Hartree–Fock method within split valence
polarized 6-31G(d) basis set using Gaussian 98 package. All calculated frequencies were
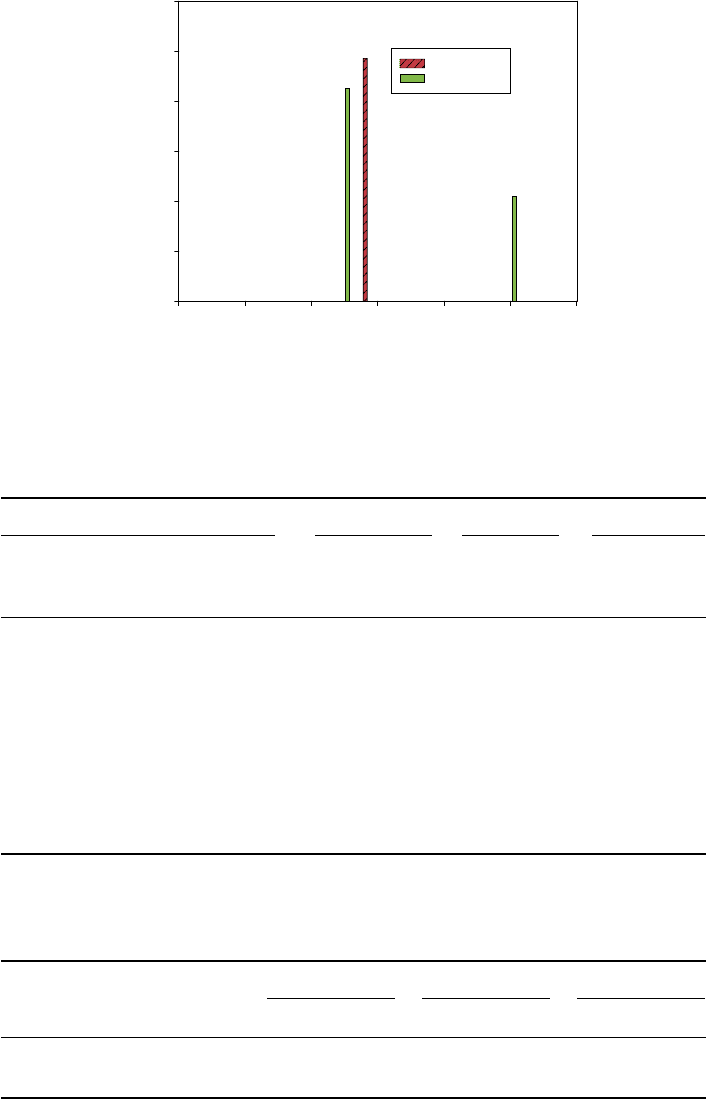
multiplied by empirical factor 0.893 to eliminate the known systematic errors. The very
far infrared (FIR) spectra (under 30 cm
−1
) of four isomers are shown in Figures 11–13.
These results illustrate that the various metastable-state conformation yields spectra
fidelity (e.g. 22 cm
−1
in 9-cis,12cm
−1
in 11-cis,26cm
−1
in 13-cis) that can be used
in defining multiple communication channels and for realizing the MS
3
approach to
expand the amount of bio-signature information. We compared our theoretical vibra-
tional frequencies with the density functional calculations of Gervasio [36], and also
with the experimental data measured by THz time-domain spectroscopy at 10 K [37]
and by FTIR spectrometer at 15 K [36]. As shown in Table 2, the frequencies pre-
dicted in our simulation are in very good agreement with the calculations of Gervasio
and comparable to experimental data above 40 cm
−1
. It is noteworthy that vibrational
modes under 40 cm
−1
exist in both simulations but are not available in experimental
70 Ying Luo et al.
Vibrational frequency (cm
–1
)
0.0
0.5
1.0
1.5
2.0
2.5
3.0
all-trans-IR
9-cis-IR
0 5 10 15 20 25 30
IR intensity (km/mol)
Figure 11 FIR spectra of 9-cis-retinal and all-trans-retinal (<30cm
−1
) [2]
Vibrational frequency (cm
–1
)
0.6
0.4
0.2
0.0
0.8
1.0
1.2
1.4
1.6
1.8
0 5 10 15 20 25 30
IR intensity (km/mol)
all-trans-IR
11-cis-IR
Figure 12 FIR spectra of 11-cis-retinal and all-trans-retinal (<30 cm
−1
) [2]
measurements. This can be explained by the experimental difficulties in the range of
40 cm
−1
and below.
This research effort also generated results for the potential energy barriers between
the ground state and metastable-state conformations (which are important for specify-
ing switching times) and the electronic excited-state energies (which are needed for
specifying the necessary optical excitation).
Calculations of the potential energy curves (PEC) in the ground-state S
0
were carried
out at both HF theory and density functional theory (DFT) levels with the split valence
polarized 6-31G(d) basis set using the Gaussian 98 package [31]. The B3LYP density
functional was used, which is Becke’s three-parameter hybrid functional using the
Lee, Yang, and Parr (LYP) correlation functional [38]. We calculated the energy of
9-cis-retinal, 11-cis-retinal, 13-cis-retinal and all-trans-retinal isomers and simulated
the rotation of all-trans-retinal around C11
==
C12, C13
==
C14, and C9
==
C10 double
Bio-molecular devices for terahertz frequency sensing 71
Vibrational frequency (cm
–1
)
0.4
0.2
0.0
0.8
0.6
1.0
1.2
0 5 10 15 20 25 30
IR intensity (km/mol)
all-trans-IR
13-cis-IR
Figure 13 FIR spectra of 13-cis-retinal and all-trans-retinal (<30 cm
−1
) [2]
Table 2 Vibrational frequencies of isolated retinal isomers in the range of FIR spectra (cm
−1
)
All-trans-retinal 9-cis-retinal 11-cis-retinal 13-cis-retinal
Ref.
[37]
exp
Ref.
[36]
exp
Ref.
[36]
cal
This
work
Ref.
[37]
exp
This
work
This work Ref.
[37]
exp
This
work
17 141 12.2 56 12.7
29 300 22.0 123 25.3
35 308 27.2 305 30.3
46.6 46 49 427 43.5 337 41.7 47.7
54.4 53 59 575 53.9 54.0 544 54.8 58.5
61.0 59.9 627 61.0
66.2 64 64.8
69.3 68 78 755 81.1 68.6 75.2
90.6 89 99 960 92.8 942 98.8
1000
Table 3 Energy barrier in the PECs of isolated retinal in the ground state (eV)
C9
==
C10 C11
==
C12 C13
==
C14
HF B3LYP HF B3LYP HF B3LYP
Barrier between 0
and 90
1.8360 1.4334 1.4878 1.1859 1.6728 1.2974
Barrier between 90
and 180
1.8578 1.4634 1.7517 1.4035 1.6946 1.3274

72 Ying Luo et al.
Torsional angle θ
11 12
(degree)
0.0
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
DFT
HF
0 20 40 60 80 100 120 140 160 180 200
Energy of molecule (eV)
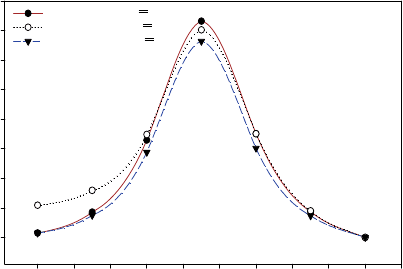
Figure 14 PECs of all-trans -retinal rotating around C11=C12 double bond calculated at HF
and DFT level
bonds. We took the dihedral angles of the corresponding double bonds
C11=C12
C13=C14
and
C9=C10
as the reaction coordinates which were fixed during the optimizations,
although all other parameters were free. The PECs of the ground state of rotation around
C11
==
C12 double bond calculated at both HF and DFT levels are shown in Figure 14.
According to our calculations, the results at the DFT level qualitatively agree with
those at the HF level although they achieve better accuracies by including electron
correlation. Also, both results predict significant barriers in the ground state that prevent
the transition from one isomer to another without external excitation.
In addition, PECs of the ground state of rotation around C9=C10, C11=C12, and
C13=C14 double bonds at DFT level are shown in Figure 15. It is clear that all-trans-
retinal has the smallest energy among all retinal isomers and is the most stable confor-
mation of retinal. 11-cis-retinal, 13-cis-retinal and 9-cis-retinal are local minima on the
PECs which means they are metastable conformations. Rotation around C9=C10 double
bond has the biggest barrier and around C13=C14 has the smallest barrier. The complete
list of barriers of the three isomerizations in the ground states are illustrated in Table 3.
To investigate the external conditions (proper light frequency) needed to initiate the
isomerization processes, we calculated the low-lying excited-states of all-trans-retinal
and its isomers. The energies of the excited states as shown in Table 4 are found within
time-dependent DFT (TDDFT) methods. For molecules with low-lying excited-states,
Table 4 Excitation energies of retinal isomers calculated with TDDFT method (eV) and OS
represents oscillator strength
9-cis-retinal 11-cis-retinal 13-cis-retinal All-trans-retinal
E(eV) OS E(eV) OS E(eV) OS E(eV) OS
S
1
3.0708 0.9110 3.0490 1.0821 3.0673 1.0771 3.0496 1.2225
S
2
3.0833 0.0006 3.0505 0 3.0991 0.0002 3.0646 0.0054
S
3
3.8894 0.5567 3.8612 0.5131 3.9118 0.5023 3.8893 0.5745
Bio-molecular devices for terahertz frequency sensing 73
rotation around C9 C10
rotation around C11 C12
rotation around C13 C14
Torsional angle θ (degree)
1.6
1.4
1.2
1.0
0.8
0.6
0.4
0.2
0.0
0 20 40 60 80 100 120 140 160 180 200
Energy of molecule (eV)
Figure 15 PECs of the ground state of all-trans-retinal rotating around C9==C10, C11==C12,
and C13==C14 double bonds at DFT level
TDDFT makes a considerable improvement over HF-based methods (like CIS) [39].
The excitation energies are similar for the corresponding states of these four isomers and
the lowest excitation energies 3.07 eV (ca. 403 nm) are in the visible region. The similar
excitation energies show that our existing simulations are not sufficient to explain the iso-
merization mechanism around different double bonds. As mentioned before, in rhodopsin
the isomerization from 11-cis-retinal to all-trans-retinal occurs but in bacteriorhodopsin
the isomerization from all-trans-retinal to 13-cis-retinal takes place. So the proper envi-
ronments, such as protein environment in this case, probably play the most important
role in determining which double bond participates in the isomerization process.
3. Retinal nanostructure arrays
Monolayers of retinal immobilized on solid substrates are of considerable importance
not only for applications as retinal-based sensors but also for investigations of the
complex behavior of retinal molecules at interfaces. By modifying one end of retinal
with a thiol linker, we could chemically graft the retinal to gold surfaces. Because
two-dimensional gold nanostructure arrays can be produced on highly oriented pyrolytic
graphite (HOPG) surfaces [40] and on semiconductor substrates [41], we can finally
fabricate two-dimensional retinal nanostructure arrays. In addition, by controlling the
two-dimensional spatial distribution of gold nanostructures including shapes and sizes,
we can control retinal molecules quantitatively.
3.1. Two-dimensional nanopatterned retinal structure
Two-dimensional nanopatterned retinal structures can be created in the following three
steps. First, connect retinal to cysteine (amino acid) which contains the thiol group by
nucleophilic addition-elimination reaction between aldehyde (C
==
O) and primary amine
(–NH
2
) as described in most organic chemistry textbooks such as [42]. The reaction is
shown in Figure 16 with the product N -retinylidene cysteine.