Moss Tom. DNA-protein interactions: principles and protocols
Подождите немного. Документ загружается.

78 Papavassiliou
digestion pattern of the DNA–protein complexes revealed on the autoradio-
graph with that of free DNA shows a band-free region (footprint) where the
bound protein(s) has prevented access of the enzyme to DNA (see Chapter 3).
In a similar analysis, the DNA is allowed to react mildly with DMS, which
methylates primarily deoxyguanosine residues and renders their phosphodiester
linkages labile under conditions of Maxam–Gilbert chemistry (see Chapter 14).
The binding of a protein(s) to a specific DNA region will result in a protection
of the corresponding bases from chemical modification (2).
The suitability of the above assays in determining the binding sequences of
proteins on DNA is hindered by several disadvantages. First, the clarity of the
footprint is highly dependent on the extent of occupancy of the binding site(s)
(i.e., a “clear” footprint is observed only if all DNA molecules are involved in
complexes). Unfortunately, this is not always easy to achieve, especially when
the concentration and/or purity of the specific binding protein(s) is not satis-
factory. Second, DNA–protein complexes formed in crude extracts may often
be heterogeneous in terms of both binding specificity and kinetic stability.
Therefore, direct footprinting in solution will not correspond to a single spe-
cies, but, instead, reflect an “integral” of the multiple equilibria operating over
the entire region of interest (i.e., the protection pattern will actually represent a
composite of more than one complex, with complexes having a very low disso-
ciation rate dominating the footprint). Finally, two different proteins that
recognize the same sequence within the probe are most likely to yield indistin-
guishable footprints. These drawbacks may be overcome by coupling treat-
ment with a footprinting reagent in solution with the electrophoretic mobility
shift assay (EMSA; also known as gel retardation assay, see Chapter 2) (3–5).
In this approach, the protein and DNA molecules are incubated together, and
the equilibrated reaction mixture is exposed to DNase I or DMS, as before. The
DNA–protein complexes are subsequently isolated from the free probe by elec-
trophoresis in a nondenaturing polyacrylamide gel. Although the negatively
charged free DNA migrates rapidly toward the anode, once it is bound by a
specific protein its mobility decreases (3,4). Following the separation of the
free and bound DNA species, the corresponding bands are cut out of the gel,
and the DNA eluted and analyzed on a sequencing gel. The region(s) of protec-
tion evident in the DNA derived from the complexed fraction, indicates the
binding site (5). Because the complexes are separated from contaminating
unbound DNA fragments, their footprints will be free of background cutting,
and thus considerably more evident. Similar considerations apply when more
than one complex can be formed on the fragment. As long as the DNA-binding
proteins differ in their molecular masses and charges, they will cause altered
electrophoretic mobilities of the corresponding complexes and, hence, differ-
ent migration in the native polyacrylamide gel. These complexes can be iso-
In Gel
OP–Cu Footprinting 79
lated and run in individual lanes on the sequencing gel. Thus, the exposure of
the binding reaction to footprinting reagents, in combination with the fraction-
ation offered by mobility shift gels, permits identification of the regions of
DNA bound by protein in different complexes, even if a low percentage of the
initial DNA molecules has been complexed.
Although one can substantially increase the sensitivity of DNase I or DMS
footprinting experiments in solution by employing the EMSA, several addi-
tional problems have still to be faced:
1. DNase I is a relatively bulky molecule (molecular weight [MW] 30,400) that
cannot cleave the DNA in the immediate vicinity of a bound protein because of
steric hindrance. As a result, the region(s) protected from cutting extends beyond
the actual protein-binding site.
2. The nonrandom nature of DNA cleavage by DNase I makes it impossible to assess
the involvement in protein binding of nucleotides that lie in an area of the frag-
ment resistant to the endonucleolytic activity of this enzyme (e.g., tracts of A and
T residues, or TpA [as opposed to ApT] dinucleotide islands scattered within or
adjacent to the binding site), so that binding sites or parts of binding sites may not
be detected.
3. The primary site of reaction of DMS with B-DNA is the N-7 atoms of guanine
bases, which are located in the major groove. Thus, those guanines in close prox-
imity with the protein will be protected from methylation. However, if a protein
primarily makes contacts with a DNA sequence in the minor groove, or if there
are no guanine residues in a major groove-binding site, DMS will not reveal
these interactions.
4. In many instances, particularly when a complex has a relatively high “off” rate,
the bound protein can dissociate from the protected DNA fragment and reassoci-
ate to other DNA fragments that have already been nicked by DNase I or modi-
fied by DMS. In this case, the DNA-cleavage pattern derived from the complexed
fraction will closely resemble that of the uncomplexed DNA, rendering it diffi-
cult to observe a footprint. The limitations imposed by the size and the sequence
or base specificity of the aforementioned footprinting reagents, as well as the
problem of protein exchange from the binding site(s) during treatment, are cir-
cumvented by merging the advantages inherent in the EMSA, with the subse-
quent exposure of the gel (hence of the resolved complexes while embedded in
the polyacrylamide matrix) to a chemical DNA-scission reagent namely the 1,10-
phenanthroline–copper ion (OP–Cu) (6).
1.1. OP–Cu as a Footprinting Agent
1.1.1. Chemistry of DNA Cleavage
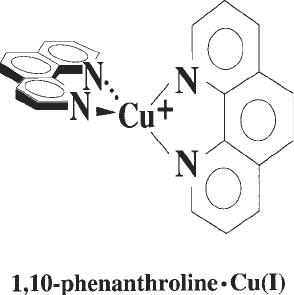
1,10 Phenanthroline–copper (Fig. 1) is an efficient chemical nuclease that
cleaves the phosphodiester backbone of nucleic acids at physiological pH and
temperature by oxidation of the deoxyribose (DNA) or ribose (RNA) moiety
80 Papavassiliou
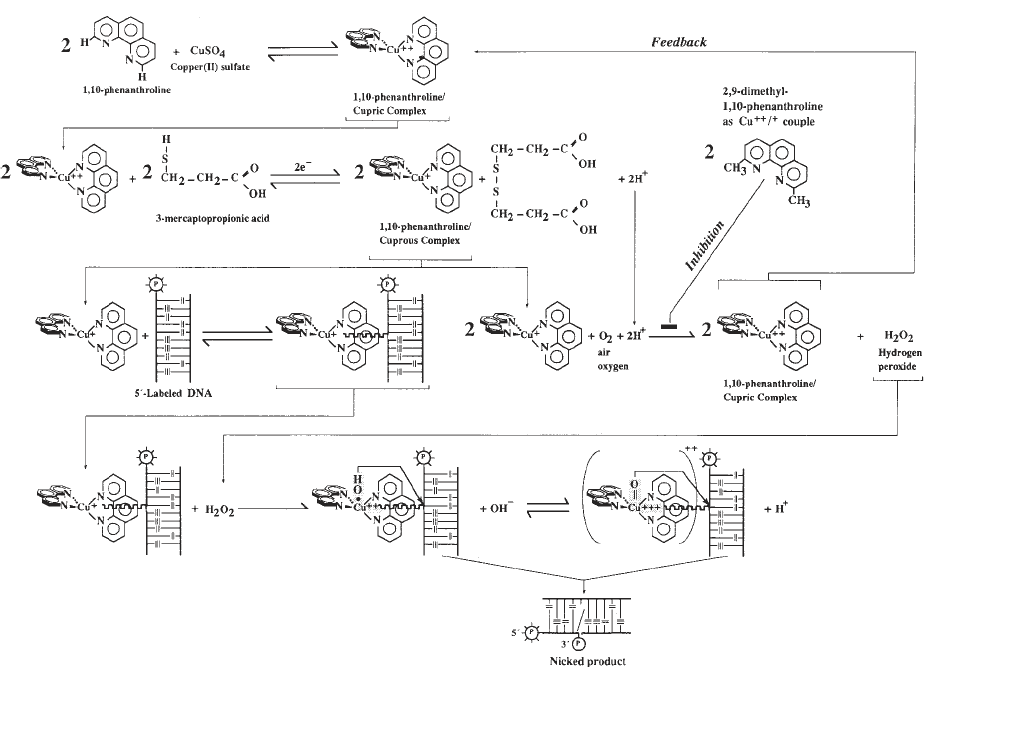
(7). The kinetic scheme of the reaction is summarized in Fig. 2. The first step is
the formation of the 1,10-phenanthroline-cupric ion coordination complex,
under conditions that favor the 2:1 stoichiometry ([OP]
2
Cu
2+
). The DNA-
scission process is initiated by adding a reducing agent, usually 3-mercapto-
propionic acid (a thiol), to the aerobic reaction mixture containing the target
DNA. Under these conditions, the 2:1 cupric complex is reduced to the 2:1
cuprous complex ([OP]
2
Cu
+
) that is, in turn, oxidized by molecular oxygen to
generate hydrogen peroxide. Hydrogen peroxide is an essential coreactant for
the chemical nuclease activity and can be generated as described above or
added exogenously (8). The tetrahedral cuprous complex, present at the steady-
state concentration defined by the experimental conditions (note the feedback
mechanism in Fig. 2), then binds reversibly to the minor groove of DNA to
form a central intermediate through which the reaction is funneled (9). The
DNA-bound cuprous complex undergoes in situ a one-electron oxidation by
hydrogen peroxide to form a short-lived, highly reactive DNA-bound copper-
oxo species that can be written either as a hydroxyl radical coordinated to the
cupric ion or as a copper-oxene structure (Fig. 2). This species then attacks the
H1'-deoxyribose protons of nucleotides, which are accessible in the minor
groove; this reaction initiates a series of reactions culminating in cleavage of
the phosphodiester backbone (9). Reaction rates at any given sequence posi-
tion depend on the stability of the intermediate formed between DNA and
(OP)
2
Cu
+
and on the orientation and proximity of the copper-oxo species rela-
tive to the C1'-deoxyribose hydrogen in the minor groove. Because both crite-
ria are met satisfactorily in B-DNA sequences, the tetrahedral cuprous complex
prefers B-DNA as its substrate. Such stereoelectronic interactions are less effi-
cient in the broad minor groove of A-DNA and not possible in Z-DNA, which
Fig. 1. Structure of 1,10-phenanthroline complexed with copper(I) ion (OP–Cu).
In Gel
OP–Cu Footprinting 81
81
Fig. 2. Schematic representation of the kinetic mechanism for the nuclease activity of 1,10-phenanthroline
–copper ion.
82 Papavassiliou
has practically no minor groove; as a result, A-DNA is cleaved at 25–33% of
the rate with which B-DNA is cleaved and Z-DNA is not cleaved at all (9).
The products of the strand-scission event include the free base, DNA frag-
ments bearing 5'- and 3'-phosphorylated termini, and the deoxyribose oxida-
tion product 5-methylene-2-furanone (10). The DNA-chain cleavage
reaction can be efficiently quenched by adding to the mixture 2,9-dimethyl-
1,10-phenanthroline (2,9-dimethyl–OP). This phenanthroline derivative can
also chelate copper ions to form a minor groove-associated cuprous complex
(thus competing with [OP]
2
Cu
+
), but the reduction potential of the Cu
2+
/Cu
+
couple is too positive to allow significant nuclease activity under normal assay
conditions (11).
1.1.2. OP–Cu Footprinting Following EMSAs
In as much as the structural and functional properties of DNA are not altered
by entrapment in a polyacrylamide gel matrix (6), the small size and the ready
diffusibility of all reaction components in solid supports permit the coupling of
OP–Cu footprinting with the EMSA to study DNA–protein interactions
(12,13). In this method, the DNA-binding reaction is performed as usual, elec-
trophoresed under established, nondenaturing conditions, and the entire mobil-
ity shift gel is immersed in a footprinting reaction mixture containing
1,10-phenanthroline, cupric ion, and 3-mercaptopropionic acid. Following the
reaction quench with 2,9-dimethyl–OP, footprints are obtained after elution of
the radioactive free and protein-bound DNA cleavage products from the
mobility shift gel and analysis on a sequencing gel (Fig. 3). Because the
nuclease activity of (OP)
2
Cu
+
produces 3'-phosphorylated and 5'-phosphory-
lated ends as cleavage products, sequencing gels can be accurately calibrated
with the Maxam–Gilbert sequencing reactions.
1.2. Advantages of OP–Cu over Other Footprinting Agents
1.2.1. General Considerations
The nuclease activity of (OP)
2
Cu
+
bears several advantages as a footprinting
reagent relative to protection analyses using DNase I or DMS as a probe. First,
the (OP)
2
Cu
+
chelate is a small molecule (compared to DNase I) that permits
cleavage closer to the edge of the DNA sequence protected by protein binding
and, therefore, a more precise definition of it. Second, because the scission
chemistry involves attack on the deoxyribose moiety, (OP)
2
Cu
+
is able to cut at
all sequence positions regardless of base. However, the intensity of cutting
(rate of cleavage) does depend on local sequence, with attack at adenines of
TAT triplets being most preferred (14; see also legend to Fig. 3). Interestingly,
a preference for C-3',5'-G steps, rather than T-3',5'-A steps, is observed at a
In Gel
OP–Cu Footprinting 83
phenanthroline to copper ratio of 1:1, which strongly favors formation of the
OPCu
+
complex (15). Nevertheless, the cutting patterns obtained with
(OP)
2
Cu
+
are usually sufficiently well-defined to identify protected regions,
even though this endonucleolytic agent exhibits some degree of sequence speci-
ficity in its rate of cleavage of naked DNA. Third, because (OP)
2
Cu
+
binds to
the minor groove of DNA, it will reveal minor-groove interactions. Because
the binding of the coordination complex should be restricted to three base
pairs, the complex is more sensitive to local, protein-induced conformational
changes than DNase I, which by possessing an extended minor groove-bind-
ing site, may be unable to sense. In this context, the complex will also detect
binding in the major groove when its approach to its minor groove-binding site
is sterically blocked or if the interaction of the protein in the major groove
alters the minor groove geometry so that the tetrahedral coordination complex
binds poorly (both being frequent features of DNA–protein interactions). Fur-
thermore, because of the difference in their respective mechanisms of cleav-
age, DNase I and (OP)
2
Cu
+
probe different aspects of the structure of a
DNA–protein complex. DNase I cleavage relies on the accessibility of a par-
ticular phosphodiester bond, and thus protection is indicative of an interaction
on the outer face of the DNA helix. In contrast, protection from (OP)
2
Cu
+
-
mediated cleavage is most likely caused by the inhibition of its binding to the
minor groove and implies that a portion of the protein occupies at least the
minor groove. Finally, in contrast to other chemical nucleases such as ferrous
EDTA (introduces single-stranded nicks in DNA through the generation of
diffusible hydroxyl radicals; see Chapter 5), the nucleolytic activity of OP–Cu
is not inhibited by glycerol, a free radical scavenger, which is present in most
protein storage buffers.
1.2.2. Benefits of OP–Cu Footprinting Within Mobility Shift Gels
The major advantage of the combined OP–Cu footprinting procedure arises
from the topography of treatment: Preformed DNA–protein complexes are
exposed to the chemical nuclease within the gel (i.e., not prior but subsequent
to an electrophoretic mobility shift experiment). This characteristic of the tech-
nique makes it ideal for protection analysis of kinetically labile complexes (16).
At least three factors account for the latter. The first is that the background
cleavage is greatly reduced by the separation of unbound DNA from the DNA–
protein complex(es) pool. The second factor is the so-called “caging effect”
(3,4). The gel matrix forms “cagelike” compartments that prevent a dissoci-
ated protein from diffusing away from the DNA, so that by enhancing
reassociation, the apparent affinity constant will be higher than the true value.
The protein could also interact with the gel matrix, thereby orienting its diffu-
sion toward reassociation. Whatever the mechanism(s), the increase in stabil-
84 Papavassiliou
84

In Gel
OP–Cu Footprinting 85
ity of the complex contributed by the gel leads to a more efficient blockage of
the access of the (OP)
2
Cu
+
chelate to the protein-binding DNA segment.
The third factor comes from the nature and site of action of the cupryl
intermediate through which the reaction is funneled, and it acts synergisti-
cally with the previous one. Because this highly reactive oxidative species is
generated near the surface of the DNA (in situ), diffusible radicals, if formed at
all, will have a short or restricted diffusive path and, therefore, will be unable
to achieve a fast equilibrium distribution along the DNA polymer. Conse-
quently, protein-binding sites exposed during multiple dissociation events will
escape the nucleolytic attack most of the time and hence remain intact.
In addition to the fact that discrete complexes with defined stoichiometries
and a wide range of kinetic stabilities can be mapped simultaneously, the in
situ OP–Cu footprinting procedure is superior to oligonucleotide-binding com-
petition assays in the analysis of multiple complexes frequently obtained in
electrophoretic mobility shift experiments employing unfractionated extract
preparations. For example, multiple retarded bands can arise from protein–
protein interactions between a non-DNA-binding transcription factor(s) and a
specific DNA-binding protein, or from two proteins binding to distinct DNA
sequences in a cooperative manner (12). Although both complexes would be
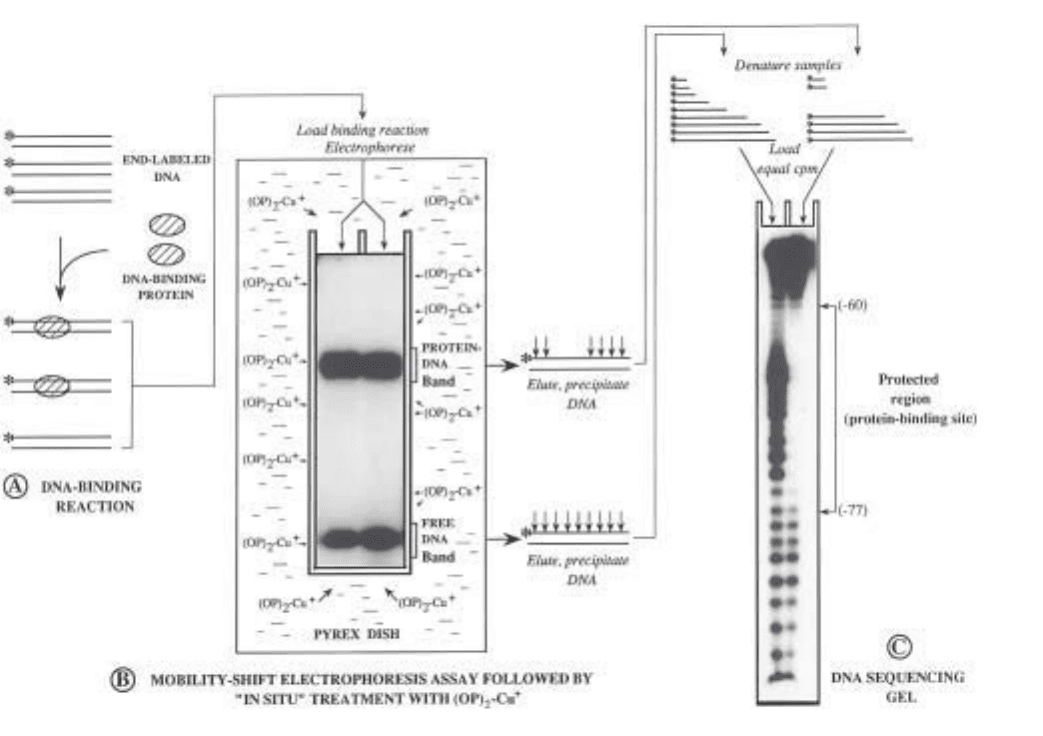
Fig. 3. (previous page) Outline of the combined electrophoretic mobility-shift/in
gel OP–Cu footprinting assay. (A) DNA restriction fragments containing a protein-
binding site(s) are labeled with
32
P at a unique end and incubated with a crude or
partially purified extract containing the DNA-binding protein(s) of interest, under
optimized binding conditions. (B) After equilibration of the DNA-binding reaction,
the free and bound DNA fragment populations are separated by electrophoresis through
a nondenaturing polyacrylamide gel; the gel is then transferred into a buffer-contain-
ing Pyrex dish, and the retarded and unretarded DNA species are exposed in situ to the
nuclease activity of (OP)
2
Cu
+
. The two DNA fractions are subsequently located by
autoradiography of the wet gel, excised and eluted from the gel matrix, precipitated,
and recovered in formamide buffer. (C) Samples are heat-denatured and equal amounts
of radioactivity from the two fractions are electrophoresed on a denaturing polyacryla-
mide gel (DNA sequencing gel) and autoradiographed. In the sample prepared from
the free-DNA band, bands will appear in the gel corresponding to positions of protein
binding. For the sample(s) prepared from the protein–DNA band(s), bands will appear
at all positions except those bound by the protein(s) (protected region). The particular
example depicts the OP–Cumapping of a DNA–protein complex formed between bac-
terially expressed LFB1 (a liver-specific transcription factor) and an oligonucleotide
bearing its binding site within the –95 to –54 region of the α1-antitrypsin promoter.
Arrowheads connected by line demarcate the footprinted site. The enhanced cleavage
observed within the protein-binding site in the free-DNA sample is the result of the
presence of repeated TA elements in this sequence (see Subheading 1.2.1.).
86 Papavassiliou
abolished by competition with oligonucleotides, these possibilities can be
readily distinguished by direct footprinting within the gel.
1.3. Additional Applications and Outlook
The nucleolytic activity of (OP)
2
Cu
+
in a polyacrylamide matrix has been
also demonstrated to be a viable means of gaining insight into the interactions
of RNA-binding proteins with their recognition sequences (17,18). Applica-
tion of OP–Cu in this context may be invaluable toward defining structural
perturbations in RNA on protein binding and mapping the binding domains of
various proteins. Because of the preferential nucleolytic activity of (OP)
2
Cu
+
toward single-stranded bulge and loop RNA regions (double-stranded stem
regions can be cut at elevated concentrations of the chemical nuclease), hyper-
sensitive sites may be obtained on footprinting an RNA–protein complex
following a gel retardation assay. Such sites would imply an unwinding of a
helical structure on protein binding or perturbations in the minor groove acces-
sibility of the bound RNA molecule.
The in gel OP–Cu footprinting methodology has already expanded the
“tool box” available to investigators wishing to explore the structure and
function relationships of nucleic acid–protein complexes, and emerging
improvements in the chemical mechanism (e.g., DNA-strand scission by the
coordination complex of OP with a non-redox-active metal) as well as future
modifications will likely make this technology even more efficient and
broadly useful.
2. Materials
2.1. Analytical and Preparative EMSA
2.1.1. Solutions
1. A variety of binding and gel buffers are commonly employed in EMSA (see Chap-
ter 2). A suitable binding buffer is 10 mM HEPES pH 7.9, 10% glycerol,
0.1 mM EDTA, 0.5 mM tetrasodium pyrophosphate, and 0.5 mM PMSF. The
most common gel buffers are Tris-glycine: 50 mM Tris, 2.5 mM EDTA, and 0.4 M
glycine; 0.5X TBE: 45 mM Tris, 45 mM boric acid, and 1 mM EDTA; Tris–acetate:
6.7 mM Tris-HCl, pH 7.5, 3.3 mM sodium acetate, and 1 mM EDTA.
2. Ammonium persulfate (10%; w/v): Weigh out 1 g of ammonium persulfate and
put it in a sterile plastic tube containing 10 mL of distilled, deionized water.
Vortex vigorously until the salt is completely dissolved. Filter through a 0.22-µm
membrane filter. This solution may be stable for a period of a few days at 4°C,
but it is recommended that you prepare it freshly for each new gel. Ammonium
persulfate is extremely destructive to tissue of the mucous membranes and upper
respiratory tract, eyes, and skin. Inhalation may be fatal. Exposure can cause
gastrointestinal disturbances and dermatitis. Wear gloves, safety glasses, respira-
In Gel
OP–Cu Footprinting 87
tor, and other protective clothing and work in a chemical fume hood. Wash thor-
oughly after handling.
3. Dye-containing binding buffer: 0.05% (w/v) bromophenol blue in 1X optimized
binding buffer (store at 4°C after filtering).
2.1.2. Reagents/Special Equipment
1. Highly purified duplex DNA fragment labeled exclusively at one of its four ends
(5' or 3'); use standard procedures for unique labeling (19). All necessary precau-
tions should be observed to minimize exposure to ionizing radiation during label-
ing and isolation of the probe. Consult the institutional environmental health and
safety office for further guidance in the appropriate use of radioactive materials.
2. Reagents employed in the optimized binding reaction.
3. (16–18) × (16–18)-cm front and back glass gel electrophoresis plates: The plates
must be absolutely clean before use. Wash them with warm soapy water; then,
holding them by the edges, rinse several times first in tap water and then in deion-
ized water. Finally, rinse with ethanol and let them air-dry. Using a pad of
Kimwipes, siliconize the inner side of the back plate with a 2% dimethyl-
dichlorosilane solution in 1,1,1-trichloroethane in a chemical fume hood (this
product is particularly toxic; gloves, safety glasses, respirator, and other protec-
tive clothing should be worn when handling it.
4. 0.3-cm spacers.
5. Electroresistant plastic tape (e.g., 3M yellow electrical tape).
6. 0.22 and 0.45-µm filters (Millipore, Bedford, MA).
7. N,N,N',N'-Tetramethylethylenediamine (TEMED; Bio-Rad, Richmond, CA).
TEMED is extremely destructive to tissue of the mucous membranes and upper
respiratory tract, eyes, and skin. Inhalation may be fatal. Prolonged contact can
cause severe irritation or burns. Wear gloves, safety glasses, respirator, and other
protective clothing and work in a chemical fume hood (TEMED is also flammable!).
Wash thoroughly after handling.
8. 3-mm gel comb with 10-mm-wide teeth.
9. 10-mL syringe and 18-gage needle.
10. 100- to 200-µL Hamilton syringe.
11. Additional reagents and equipment: Powdered acrylamide and N,N'-methylene–
bis-acrylamide (Bio-Rad); plenty of binder clamps (fold-back spring clips); razor
blades; polyacrylamide gel electrophoresis apparatus; constant current power
supply; peristaltic pump for recirculating electrophoresis buffer (if required);
siliconized 1.5-mL Eppendorf microcentrifuge tubes; spatula. Acrylamide and
N,N'-methylene–bis-acrylamide are potent neurotoxins and are absorbed
through the skin. Their effects are cumulative. Wear gloves and a face mask
when weighing these substances and when handling solutions containing them.
Although polyacrylamide is considered to be nontoxic, it should be handled
with care because of the possibility that it might contain small quantities of
unpolymerized acrylamide.