Jackson S.D., Hargreaves J.S.J. Metal Oxide Catalysis
Подождите немного. Документ загружается.


254 6 Photoelectron Spectroscopy of Catalytic Oxide Materials
The chemical shift of the bulk contribution to the oxygen 1s spectrum is domi-
nated by the valence state mixing between oxygen and metal centers. The electro-
negative oxygen tends to abstract electron density from the metal centers, with
resulting high local ground state electron densities at oxygen and low binding
energies of about 528.5 eV for later transition metal systems. In early transition
metal systems, the charge redistribution is less effective, shifting the binding
energy up to 531.5 eV with a large family of binding - energy values at about 530.5 eV.
Main group elements are even less effective charge - donating partners, giving rise
to positions of up to 533 eV for water and silicon oxides. In this latter group, the
geometry of the bonding has a substantial infl uence on the binding energy, making
XPS a structure - sensitive tool. In d - block metal oxides, only major changes in the
d – p rehybridization (for example in copper oxides with 529.5 eV for CuO and
530.5 eV for Cu
2
O) exert a signifi cant infl uence, otherwise the position is rather
insensitive to, for example, the formal oxidation state of metal center. Compila-
tions of such data with a chemically broad span of samples are available [17, 34,
35] .
The interpretation of the oxygen contribution of the terminating layer to the O
1s spectrum should follow different considerations. It is known that oxides termi-
nate by very strong structural relaxations [36 – 41] , making the bulk chemical
bonding of oxygen a very poor approximation of their surface. In brief, oxides ter-
minate in their most stable form by creating formal oxygen – metal double bond
groups ( “ - yl structures ” ) also for metals where such - yl structures are not known
in the form of bulk compounds. Such terminations are stable and non - reactive,
requiring defect sites for adsorption and catalysis. The nature of defects can be
the incorporation of hydroxyl groups originally thought to be the main terminating
species [42] , but many other confi gurations of weakly held atomic or molecular
oxygen [40, 43 – 47] are also under debate. For the present consideration it follows
that all oxides should exhibit a low - energy contribution, situated below 530 eV for
stable terminations, and further contributions above 531 eV characterizing the
defect oxygen species, from which a discrimination between atomic species and
hydroxyl groups may be very diffi cult in the absence of valence band spectra, where
they yield single peak and dual peak structures that allow discrimination.
In practice, asymmetric O 1s peaks are often found for oxide systems, and these
fi nd their explanation in the coexistence of a surface spectrum and a bulk spec-
trum. The contribution of the surface spectrum for polar systems and in the pres-
ence of water strongly bound via hydrogen bridges (and thus not pumped away
in a normal XPS system without thermal desorption) may then be larger than the
estimated 15% and could amount to a clearly visible structure. The contribution
of the - yl species, however, is frequently not easily detectable, as its abundance is
strictly limited to a maximum of one monolayer, being sub - stoichiometric with
respect to a close packed metal layer as it saturates two dangling coordinations per
oxygen atom.
Efforts to separate the oxygen 1s spectrum into bulk and surface components
are desirable, as the description of chemical reactivity requires a description of the
terminating oxide structure. In cases where well characterized surface - science

6.3 Case Studies 255
model compounds are available [36, 37, 48 – 54] one can justify the fi tting of a broad
spectrum to different dominating and minor components, using the spectral
parameters of the model systems to account for the bulk and the - yl contributions
and assigning the unexplained part of the spectral weight to defects and adsorbate
features. In the majority of the other cases, one may use in situ thermal desorption
and/or re - oxidation as means to justify the splitting of the convoluted oxygen PES
into bulk and surface contributions. Care must be taken not to choose such severe
conditions of surface modifi cation that the bulk becomes affected. This can become
a matter of considerable experimental effort, as many oxides (in particular early
transition metal oxides) do exhibit pronounced tendencies to form complex sub -
oxides [55 – 59] . It is, however, almost never right to assume one type of oxygen
species to be suffi cient to describe the bulk and the surface of an oxide system.
If the separation of the spectrum into bulk and surface contributions was suc-
cessful, then an estimation of the chemical reactivity can be given using the ratio-
nale as a basis that the reactivity will be nucleophilic for low binding - energy species
and electrophilic for species with high binding energies (above 531 eV). This
assignment of nomenclature [60] refers to the reactivity of oxygen with respect to
C
−
H bonds, representing a very common case of the reactivity required in hydro-
carbon transformation (partial and deep oxidation). The suggestions given here
should serve as guidelines rather than as strict rules for assigning spectra. A large
number of peculiarities, such as those mentioned above, of the system under study
need to be considered, as well as the strict and conclusive elimination of binding -
energy scale artefacts already described. Only then can peak deconvolution be
made with the degree of chemical resolution required for reactivity assignments
without simply converting wishful thoughts into unresolved lines.
6.3
Case Studies
6.3.1
Applications of XPS to Vanadium Oxides
Metal oxides play an important role in many fi elds of our society, one of which is
heterogeneous catalysis. These materials can be applied as carriers of an active
component, and also as the active material itself. We have selected vanadium – and
vanadium - containing – oxides as our topic, because they are involved in many cata-
lytic processes, most likely due to their versatile electronic structure. Vanadium,
with its valence atomic electron confi guration of 3d
3
4s
2
, forms different types of
oxides ranging from insulators to metal, depending on the valence shell confi gura-
tion and temperature. Their electronic structure was comprehensively studied by
several research groups [50, 61 – 67] . Zimmermann and coworkers [63] demon-
strated a strong hybridization between the V 3d and O 2p orbitals, and owing to
the covalency in bonding, such early transition metal oxides (e.g. V
x
O
y
) cannot be
considered as simple Mott – Hubbard compounds. Density - functional theory ( DTF )

256 6 Photoelectron Spectroscopy of Catalytic Oxide Materials
calculations [64, 67] confi rmed the strong hybridization of the valence oxygen and
vanadium orbitals in V
2
O
5
, and related the distortion of the VO
6
octahedra to the
unique electronic structure of the conduction band. A recent DFT cluster study
[50] mimicking V
2
O
5
has clearly indicated that the local charges (Mulliken) of the
different cluster atoms are much smaller than formal valence charges (V
+1.4
; O
− 0.26
,
O
− 0.58
, O
− 0.78
for the three different lattice O positions), in line with the suggested
covalent bonding contribution. DFT calculations on other oxides reported similar
discrepancies between partial and formal charges [68 – 71] . It is well known among
quantum chemists, and we would also stress, that although Mulliken charges and
formal valence charges may yield the same qualitative picture, they cannot be
compared on a quantitative basis. Formal valence charges may be useful in certain
cases, but if considered as a universal tool, they can easily lead to erroneous con-
clusions and reaction models, as often observed in the heterogeneous catalysis
community. In what follows, we will use integer valence charges (e.g. V
5+
) to rep-
resent formal oxidation states, while fractional numbers (e.g. V
+1.4
) will indicate
local charges calculated by DFT methods.
The electronic structure of vanadium oxides is crucial to their reactivity. Deter-
mination of even the formal oxidation state of vanadium from XPS can, however,
be non - trivial, owing to a variety of factors. Firstly, examination of binary vanadium
oxides has highlighted that there appear to be differences in binding - energy posi-
tions and FWHM for single crystal and powder samples (see Table 6.1 ). In general,
the binding energy quoted for V
2
O
5
is quite consistent, ranging from 516.9 to
517.2 eV. However, Table 6.1 shows that the ranges of energies given for V
4+
and
V
3+
in the literature are considerably wider, and in some cases overlap. This can
be for a number of reasons, including variations in equipment/analyzer, surface
cleaning method, background and satellite subtraction and subsequent calibration
of the binding - energy scale. A relatively reliable binding - energy calibration method,
at least in the case of reduced oxides, may be to use the band gap transition of the
V 3d peak in the valence band [63] . Additionally, the widths of V 2p core levels
increase from V
2
O
5
to open valence VO
2
and to V
2
O
3
. This behavior is due to the
increasing number of available multiplet confi gurations in the corresponding
photoelectron fi nal states, that is, non - resolved multiplet splitting occurs in V 2p
core levels of lower valence vanadium oxides. This effect is also observed in the
mixed valence vanadium oxides such as V
6
O
13
(V
2 n
O
5 n − 2
, Wadsley phase) or V
4
O
7
and V
3
O
5
(V
n
O
2 n − 1
, Magn é li phase). An XPS study of these phases [74] has shown
similarities to the studies of V
2
O
5
, VO
2
and V
2
O
3
, where strong hybridization
between the O 2p and V 3d states is observed. The broad nature of the V 2p peak
of the mixed oxide phases was attributed to the mixture of oxidation states present
as well as to surface defects.
Further diffi culties in measuring vanadium oxides arise from reliable prepara-
tion of reference compounds and reduction of vanadium caused by UHV condi-
tions and beam damage [78, 79] . Hence, it is advantageous to confi rm the phases
present by additional analytical techniques.
As expected, the diffi culties in relating binding energies to formal oxidation
states of vanadium becomes more complex in the case of supported vanadium

6.3 Case Studies 257
oxide catalysts (see Table 6.2 ). Again, accurate calibration of the binding - energy
scale is essential as, in the case of the catalysts shown, there is a difference of
∼ 0.5 eV in the calibration energy chosen for the C 1s peak alone. As the catalyst
support is normally an insulating material, charge compensation is made by cali-
brating the binding - energy scale to a known value. In many cases the choice of
value (support peak or carbon) can cause discrepancies between quoted binding
energy values. Achieving an accurate peak fi t of the V 2p
3/2
region ideally requires
prior knowledge of the position and FWHM of each oxidation state, as determined
from reference compounds. However, Tables 6.1 and 6.2 highlight the diffi culties
surrounding this simple strategy.
Perhaps the starting place for investigations of supported metal oxide catalysts
by XPS should be the examination of well characterized model systems. Several
authors have investigated V
x
O
y
fi lms supported on a number of metal substrates
Table 6.1 XPS binding energy positions of a selection of V
x
O
y
materials from literature.
Sample Binding Energy of V2p
3/2
and O1s/eV (FWHM)
a)
E
B
Calibration Reference
V
5+
V
4+
V
3+
V
0
O1s
V
x
O
y
on V foil 516.9 515.8 – – – Au 4f
7/2
[72]
V
2
O
3
(001) on
W(100) and
Au(111)
517.15 – 515.15 –
∼ 530
W 4f
7/2
[52]
V
x
O
y
on V 517.2 515.8 515.2 512.4 – Au 4f
7/2
84.0 eV [73]
Single crystals
V
2
O
5
, VO
2
,
V
2
O
3
, V (foil)
516.9 (1.6) 516.2 (3.2) 515.7 (4.2) 512.4 (2.0) 529.8 – 530.1 F.E. [61]
V
2
O
5
, VO
2
,
V
2
O
3
, V
6
O
13
,
V
4
O
7
, V
3
O
5
517.2 (1.2) 516 (1.95) 515.85 512.2 – C 1s 285 eV [74]
Powders
V
2
O
5
, VO
2
, V
(foil)
516.4 (3.0) 516.1 (3.4) – 512.7 529.8 – 530.0 C 1s 284.6 eV [75]
V
2
O
5
(s.c.),
V
6
O
13
, VO
2
,
V
2
O
3
517.0 (1.3) 515.65 (4.0) 515.1 (4.8) – 529.8 – 530.0 O 1s
529.8/530 eV
[76]
V
2
O
5
, VO
2
,
V
2
O
3
517.2 516.0 514.0 – – C 1s 284.8 eV [77]
a FWHM in brackets if given in text.

258 6 Photoelectron Spectroscopy of Catalytic Oxide Materials
[36, 52, 72, 73, 88, 89] . The benefi ts of such studies are that in the case of insulat-
ing oxides, charging is reduced owing to the enhanced conductivity of the thin
fi lm in contact with a metal substrate. Nevertheless, even XPS spectra from model
systems can prove challenging to interpret. It has been reported that satellite peaks
from V 2p may have to be taken into account when fi tting peaks to the V 2p enve-
lope [52, 63] . Dupuis and coworkers [52] investigated V
2
O
3
on W(110) and used
two peaks in their description of the V 2p
3/2
curve (Figure 6.2 ). They attributed the
higher energy feature (517.5 eV) to vanadyl groups with a formal oxidation state
of 5+ and the lower peak (515.15 eV) to bulk V
2
O
3
. This assignment was aided by
angle - resolved depth profi ling, which showed an enhancement of the peak attrib-
uted to vanadyl groups closer to the surface. Although the peak position is in good
agreement with that of V
5+
, the FWHM is considerably broader than that of the
V
2
O
5
crystal shown for comparison [52] . Additionally, broadening of the O 1s peak
Table 6.2 XPS binding energy positions of deconvoluted V 2p
3/2
peaks of V
x
O
y
/support catalysts from literature
Sample Binding energy of V2p
3/2
and O1s (eV) (FWHM)
a)
E
B
Calibration Reference
V
5+
V
4+
V
3+
V? O1s
V
x
O
y
/ γ - Al
2
O
3
517.2 –
517.3
– 515.5 –
515.8
Al 2p 74.5 eV
[80]
b)
V
2
O
5
, V - Al - O
catalyst
517.0 515.75 515.2
∼ 531
C 1s 285 eV
[81]
VPO,
V
x
O
y
/ γ - Al
2
O
3
518.0 516.9 –
V
< 3+
515.1
C 1 s
284.5 eV,
Al 2p 74.5 eV
[79]
V
x
O
y
/ γ - Al
2
O
3
518.0 516.9 – V 2p of
VOPO
4
at
518 eV
[82]
V
x
O
y
/Al
2
O
3
517.4 – 517.6 516.4 – 516.5 515.8 – 515.9 Al 2p 74.5 eV
[83]
V
x
O
y
/Al
2
O
3
- ZrO
2
518.1 – 2
(1.8 – 2.0)
– – 531.8 – 532 C 1s 284.6 eV
[84]
V
x
O
y
/SBA - 15 517.3 (2.1) – 515.9 (2.1)
518.7
c)
(2.1)
– Si 2p
103.6 eV
[85, 86]
V
x
O
y
/SBA - 15 517.1 516.1 – 532.8 – 533.0 C 1s 284.6
[87]
a FWHM in brackets if given in text.
b Eberhardt and coworkers used principal component analysis and iterative transformation factor
analysis to determine number of components and position.
c High E
B
caused by fi nal state effects; due to presence of small conducting particles on insulating
substrate (see references for further details).
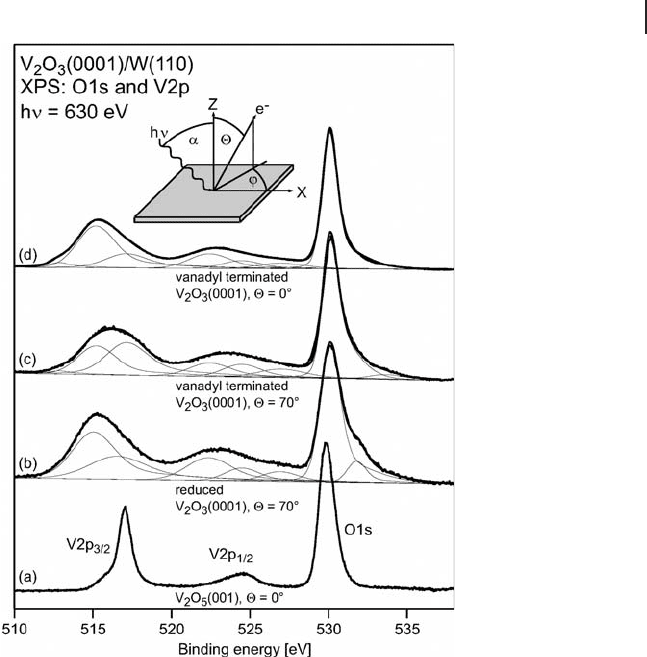
6.3 Case Studies 259
was observed, which may be due to the formation of hydroxyl groups. Dupuis and
colleagues suggested that in addition to oxidation state, screening effects can have
an infl uence on the observed binding energy, the effect of which is greater for
bulk atoms. The same group reports a correlation between intensity of the V 3d
contribution and increasing vanadium reduction [36, 52] . Studies of similar
systems [36, 88, 89] under various conditions – hydrated, reduced, oxidized –
confi rm that combined analysis of the core level and valence band XPS provides
greater information about changes in the vanadium oxide ’ s surface. The presence
of a more oxidized surface of vanadium oxide fi lms compared with deeper layers
was confi rmed by Alov and coworkers [73] . They used XPS and detected a higher
ratio of V
5+
and V
4+
on the outer part of the fi lm, whereas lower oxidation states
were concentrated at greater depths.
Fresh and post - reaction supported vanadium oxide catalysts have been exam-
ined by several authors (references and E
B
values of some examples are given in
Figure 6.2 V 2p and O 1s region of V
2
O
3
(0001) on W(110)
in comparison with V
2
O
5
according to Dupuis and coworkers
[52] . The spectra indicate that the V
2
O
3
(0001) surface is
terminated with vanadyl groups.

260 6 Photoelectron Spectroscopy of Catalytic Oxide Materials
Table 6.2 ). In general, XPS of “ fresh ” catalysts shows mainly oxidized vanadium
(V
5+
). After reduction treatments, a decrease in oxidation state is observed. However,
the extent of reduction depends on a number of factors, such as reduction tem-
perature, reducing agent and partial pressure of the reducing gas, as well as the
method used to transfer the reduced sample to the measurement chamber. Ideally,
contact with air should be minimized, or excluded if possible, to prevent re - oxida-
tion of the catalyst when the sample is transferred from a reactor to the UHV
measurement chamber. This problem can be circumvented by in situ instrumenta-
tion as discussed later.
In the following sections, we present a critical overview of the application of
X - ray photoelectron spectroscopy in vanadium - oxide related catalytic literature
(including dehydrogenation, oxidative dehydrogenation and selective oxidation
processes). Our focus will concentrate on the strengths and weaknesses of the
method and will explore what in situ experimentation can contribute to this
fi eld.
6.3.2
Direct and Oxidative Dehydrogenation
Removal of hydrogen from a hydrocarbon to form its unsaturated derivative is an
important step towards the formation of chemical feedstocks. Unsaturated hydro-
carbons are often more reactive and in high demand, hence the process is of great
value within industry. An additional benefi t is the formation of hydrogen, which
if separated from the products would prove to be highly valuable. Dehydrogenation
is usually an endothermic reaction, requiring high temperature and low pressure
to ensure a suffi cient yield of product. The exception is for dehydrogenation of
compounds such as cyclohexane where the formation of more stable aromatic
derivatives creates more favorable thermodynamics. Fortunately, the thermody-
namics of the reaction can be enhanced by addition of oxygen (oxidative dehydro-
genation). In this case water is formed as a product, resulting in a thermodynamically
favorable reaction. The major goal within the dehydrogenation reaction is to fi nd
catalysts that are highly selective to the required product while limiting carbon
deposition, which can lead to catalyst poisoning. The majority of processes for the
dehydrogenation of light alkanes use catalysts containing chromia or platinum
supported on alumina [90 – 92] . However supported vanadium oxides have also
shown applicability for the catalysis of oxidative dehydrogenation [93 – 96] and
dehydrogenation [83, 97 – 99] of light alkanes.
6.3.2.1 Selective Dehydrogenation of n - Butane over V
x
O
y
/Alumina
In each of the studies described so far, XPS was measured under UHV conditions.
However, previous studies of oxide catalysts [100] have shown that by using a
specially designed high - pressure in situ XPS apparatus, XP spectra can be mea-
sured under reaction conditions in the millibar pressure range. This technique
was applied to a selection of V
x
O
y
/ δ - alumina catalysts (1 – 8 wt% V) to determine
their electronic structure under oxidative and reaction ( n - butane) atmospheres
6.3 Case Studies 261
[101] . Initially the “ fresh ” catalysts were examined, in an atmosphere of 0.5 mbar
of oxygen at elevated temperature (623 K). From XPS, the predominant vanadium
species was V
5+
, independently of the vanadium loading (1, 3.5 and 8 wt% V on
alumina). Typically less than 4% of reduced species was observed under these
conditions. This is in agreement with other measurements of “ fresh ” catalysts as,
for example, Harlin and coworkers [83] reported that their calcined catalyst con-
tained 100% V
5+
, according to conventional XPS. In contrast, several authors [82]
suggest a higher ratio of reduced vanadium in fresh catalysts, but this may be due
to the absence of any oxidative pre - treatment or to damage caused by beam or
UHV conditions. By measuring XPS in an oxygen atmosphere and at elevated
temperature, as in the work of the present authors, both reduction and charging
of insulating samples are reduced compared with conventional XPS systems.
Although in our case the oxidation state remains unchanged with loading, it is
likely according to the literature [102, 103] that the structures of vanadium oxide
on the alumina surface vary greatly. By using the complementary technique of
XAS (Section 6.2.2.2 ), information about not only the oxidation state but also the
local environment of the element under investigation can be determined. The
vanadium L
3
edge was measured for each of the three catalysts, under the same
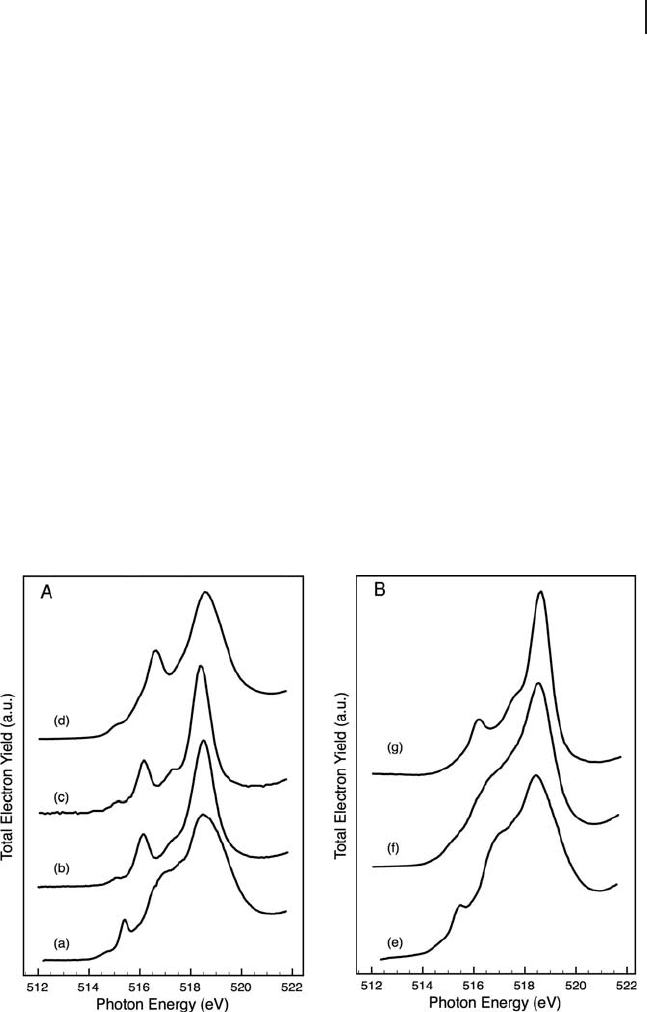
conditions as for XPS, as shown in Figure 6.3 . This edge is due to electronic
transitions from vanadium 2p
3/2
to 3d. The results were compared with a series of
Figure 6.3 V L
3
NEXAFS edge of (A) reference materials:
(a) V
2
O
5
single crystal, (b) Mg
2
V
2
O
7
(c) Mg
3
V
2
O
8
(d) MgV
2
O
6
and (B) fresh catalysts: (e) 8% V/alumina, (f) 3.5% V/alumina
and (g) 1% V/alumina. All spectra were measured in an
oxygen atmosphere (0.5 mbar) at 623 K and an angle of 55 ° .

262 6 Photoelectron Spectroscopy of Catalytic Oxide Materials
reference compounds as shown in Figure 6.3 A. The spectra shown in the fi gure
represent a selection of possible vanadium - oxygen linkages. Comparison of the
spectra indicate that 8% V/alumina corresponds well with the spectrum of crystal-
line V
2
O
5
. The low - loading catalyst (1% V/alumina) corresponds well with the
Mg
3
V
2
O
8
(monomeric) and the Mg
2
V
2
O
7
(dimeric) compounds, whereas the 3.5%
V/alumina catalyst did not appear to match any specifi c reference compound.
Although a stable reference material for the polyvanadate structure was not avail-
able, from the appearance of the vanadium L
3
edge of the 3.5% V/alumina catalyst,
it is likely that a mixture of species was present, possibly including V
2
O
5
crystallites
and polyvanadates as suggested from (UV - )Raman spectroscopy of the same cata-
lysts [103] . Owing to a high degree of complexity, theoretical calculations to defi ne
the origin of the features of the vanadium L
2,3
edge are limited. De Fransco and
coworkers [104] have presented a density - functional investigation of V
2
O
5
. Although
able to calculate theoretical absorption K edges that are similar to experimental
results, vanadium L - edge calculations still contained discrepancies from experi-
ment. In different studies [105, 106] , fi rst principles multi - electron calculations of
VO
2
and V
2
O
3
were found to fi t well to experimental fi ndings, although there was
still some disagreement with the V
2
O
5
system. Therefore, detailed calculations of
the vanadium L edge of magnesium vanadate compounds would be necessary
to relate the fi ne structure of the electronic transitions to the structure of the
compounds.
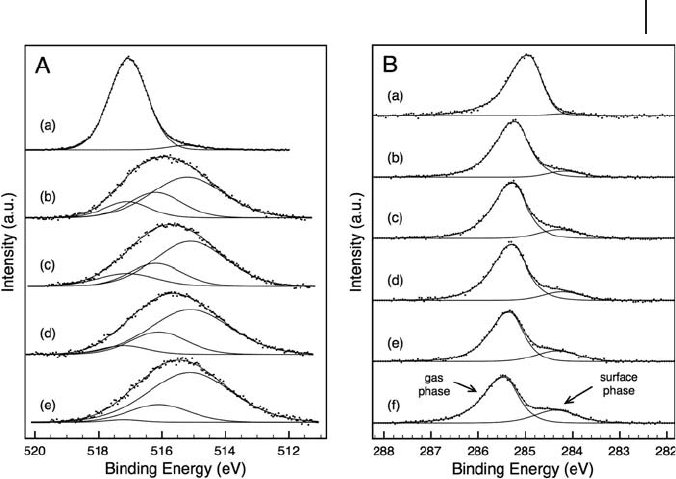
Additionally, the catalysts were examined under reaction conditions. The results
of XPS analysis of an 8% V/alumina catalyst under n - butane dehydrogenation
conditions can be seen in Figure 6.4 . During the dehydrogenation reaction in n -
butane (0.4 mbar) and at a reaction temperature of 723 K, the formal oxidation state
of vanadium was greatly reduced from the initial mixed oxidation state of 5+/4+
after O
2
/H
2
pre - treatment. The broadening of the V 2p
3/2
peak and the position of
the lower binding energy component is in agreement with the fi ndings of Dupuis
and coworkers [52] for V
3+
. One group reports the presence of only V
5+
and V
3+
on
a vanadium/alumina catalyst as determined by statistical methods [80] . In that
study , both the number of major components and their peak position were deter-
mined by principal component analysis and iterative transformation factor analy-
sis. However, as shown in Tables 6.1 and 6.2 , various authors report a value in
between that of V
5+
and V
3+
, which they assign to VO
2
or a V
4+
species. In our case
it is reasonable to fi t the V 2p envelope with three peaks representing V
5+
, V
4+
and
V
3+
(see Figure 6.4 A). Adsorption studies of probe molecules (e.g. NO or CO) also
suggest the presence of vanadium species with formal oxidation states of 3, 4 and
5+ [107, 108] . This is in agreement with the work of Harlin and coworkers [83] ,
who proposed that the active center for the dehydrogenation reaction (of n - butane
and i - butane at 853 K) is as a result of surface vacancies due to V
3+
and V
4+
at the
catalyst surface. They also suggest that reduction of vanadium is due to reaction
not only with the feedstock but also with the reaction products (butenes, butadiene
and with small amounts of cracked products). In addition to surface reduction,
these lines broaden with lower oxidation state, owing to multiplet splitting.
However, it can also be seen that the FWHM of the 5+ component increases. This
6.3 Case Studies 263
effect can be seen in the V 2p fi ttings of Dupuis and coworkers [52] , hence it is
not an effect restricted to powder samples. Hess and coworkers [85, 86] observed
an additional feature in their XPS of V
x
O
y
/SBA - 15 samples, at even higher binding
energy than expected for V
5+
. This was attributed to charging in the fi nal state by
the presence of small particles on an insulating support. Hence, the broadening
of the V
5+
contribution may have a fi nal state origin or it could be due to the pres-
ence of neighboring reduced atoms.
Concomitantly to the XPS measurements, on - line mass spectrometry shows the
main products as butenes, butadiene and, to a lesser extent, benzene. Although
this study was performed at reduced pressure, reaction products were similar to
those found in high - pressure (1 bar) studies [83, 97] . (Previous studies of n - butane
dehydrogenation have also detected benzene in the product stream [83] .) The
combination of low partial pressure of feed with parallel surface monitoring pro-
vides a unique tool to follow the processes occurring during the initial period of
reaction/deactivation in “ slow - motion. ” Indeed, even under our low - pressure con-
ditions, deactivation of the catalyst and deposition of carbon were observed. Owing
to these processes, regeneration cycles were performed by treating the catalyst fi rst
in oxygen then in hydrogen at 723 K. After each regeneration, the activity of the
Figure 6.4 (A) XP spectra of V 2p
3/2
region of 8% V/alumina
in (a) 0.5 mbar oxygen and 0.4 mbar n - butane (723 K),
after (b) 53 min, (c) 87 min, (d) 129 min and (e) 186 min.
(B) XPS of C1s region during reaction in n - butane (0.4 mbar,
723 K) after (a) 60 min, (b) 96 min, (c) 114 min, (d) 118 min,
(e) 134 min and (f) 174 min in the reaction mixture.