Drake G.W.F. (editor) Handbook of Atomic, Molecular, and Optical Physics
Подождите немного. Документ загружается.

Molecular Structure 31.2 Characterization of Potential Energy Surfaces 473
function for the ith helium atom in the natural orbital
basis [31.61] can be written as Ψ
i
=[1+ T
2
(i)]φ
0
(i),so
that the N atom electronic wave function becomes
Ψ =
i
Ψ
i
=
i
[1+ T
2
(i)]Φ
0
=
exp
i
T
2
(i)
Φ
0
= exp(T
2
)Φ
0
(31.29)
Thus this exponential type of solution scales properly
with the number of particles.
To obtain the amplitudes t,defineH
N
= H
0
−
Φ
0
|H
0
|Φ
0
, E
0−CC
I
= ∆E
0−CC
I
+Φ
0
|H
0
|Φ
0
≡
∆E
0−CC
I
+ E
0−0
I
, P =|Φ
0
Φ
0
| and Q = 1 − P. Then,
inserting (31.25–31.28)into(31.5)givestheenergy
(31.30) and amplitude (31.31) equations, which define
the coupled cluster approach:
∆E
0−CC
I
=Φ
0
|
˜
H
N
|Φ
0
, (31.30)
Φ
abc···
ijk···
|
˜
H
N
|Φ
0
=0forallΦ
abc···
ijk···
, (31.31)
where
˜
H
N
= exp(−T )H
N
exp(T)
=−E
0−0
I
+ exp(−T )H
0
exp(T ). (31.32)
To appreciate the nature of these equations, con-
sider the approximation T = T
2
constructed from SCF
orbitals [31.36, 59], referred to as the coupled cluster
doubles (CCD) level. At this level the energy equation
becomes
E
0−CCD
I
=Φ
0
| exp(−T
2
)H
0
exp(T
2
)|Φ
0
=Φ
0
|H
0
T
2
|Φ
0
+E
0−0
I
= E
0−0
I
+
1
4
i, j,r,s
Φ
0
H
0
a
†
r
a
†
s
a
i
a
j
Φ
0
t
rs
ij
= E
0−0
I
+
1
4
i, j,r,s
(rs||ij )t
rs
ij
(31.33)
for i, j occupied and r, s virtual orbitals. The amplitude
equations become
Φ
rs
ij
| exp(−T
2
)H
0
exp(T
2
)|Φ
0
=Φ
rs
ij
|
1− T
2
+
1
2
T
2
2
H
0
×
1+ T
2
+
1
2
T
2
2
|Φ
0
= 0
(31.34)
which reduces (after considerable commutator alge-
bra) [31.36]to
(λ
r
+ λ
s
− λ
i
− λ
j
)t
rs
ij
=
− (rs||ij ) −
p>q
(rs||pq)t
pq
ij
−
k>l
(kl||ij)t
rs
kl
+
k, p
(ks|| jp)t
rp
ik
− (kr|| jp)t
sp
ik
− (ks||ip)t
rp
jk
+ (kr||ip)t
sp
jk
−
k>l ; p>q
(kl|| pq)
t
pq
ij
t
rs
kl
− 2
t
rp
ij
t
sq
kl
+ t
sq
ij
t
rp
kl
− 2
t
rs
ik
t
pq
jl
+ t
pq
ik
t
rs
jl
+ 4
t
rp
ik
t
sq
jl
+ t
sq
ik
t
rp
jl
.
(31.35)
The solution to (31.35) is obtained iteratively. The
first iteration gives t
rs
ij
=−(rs||ij )/(λ
r
+ λ
s
− λ
i
− λ
j
),
which when inserted into (31.33)gives(31.24), i. e.,
the MP2 result. If the quadratic terms in (31.35)are
neglected, then the second iteration gives the third or-
der Møller–Plesset (MP3) result [31.36]. The result of
iterating (31.35) to convergence gives the CCD result.
Of the levels of coupled cluster treatments in current
use, those including T
1
, T
2
, T
3
,andT
4
in (31.25) provide
the most reliable results [31.62–66].
Multireference Methods
Single reference methods provide probably the most
powerful tools for treating near-equilibrium properties
of ground electronic state systems. In other instances,
such as the study of electronically excited states, de-
termination of global potential energy surfaces and for
systems with multiple open shells such as diradicals,
multireference techniques are found to be extremely use-
ful. In the multireference techniques discussed below,
the wave function is written as
Ψ
0−MRF
I
(r; R) =
α
c
I
α
ψ
α
(r; R) (31.36)
where ψ
α
(r; R) is a CSF. This expansion is usually
referred to as a configuration interaction (CI) expan-
sion, and the coefficients c
I
(R) are referred to as CI
coefficients [31.42]. Since (31.36) does not involve the
exponential ansatz, it is not automatically size extensive.
In the description of these wave functions, it is useful to
generalize the notion of occupied and virtual orbitals to
inactive, active, and virtual orbitals, where, referring to
the CSF’s defining the reference space, inactive orbitals
are fully occupied in all CSFs, virtual orbitals are not oc-
Part C 31.2

474 Part C Molecules
cupied in any CSF and the active orbitals are (partially)
occupied in at least one CSF.
Most multireference techniques begin with the deter-
mination of a multiconfigurational self-consistent field
(MCSCF) wave function or state averaged MCSCF
(SA-MCSCF) wave function [31.50–53, 67]. This ap-
proach is capable of describing the internal or static
correlation energy, the part of the correlation energy that
leads to sizeable separation of two electrons in a pair,
and near-degeneracy effects. A particularly robust type
of MCSCF wave function, the complete active space
(CAS) [31.68–70] wave function, includes in (31.36)all
CSFs arising from the distribution of the available elec-
trons among the active orbitals. Note that a CAS wave
function is size consistent.
The remaining part of the correlation energy, the dy-
namic correlation, describes the two-electron cusp, i. e.,
the regions of space for which two electrons experience
the singularity in the Coulomb potential. Empirically,
it has been shown [31.71] that wave functions cap-
able of providing chemically accurate descriptions of
the dynamic correlation can be obtained by augmenting
the MCSCF or reference wave function with all CSFs
that differ by at most two molecular orbitals, double
excitations (31.28) from those of the reference space.
Such wave functions are generally referred to as mul-
tireference single and double excitation configuration
interaction (MR-SDCI) wave functions. First (second)
order wave functions [31.72] include all single (sin-
gle and double) excitations relative to a CAS reference
space.
Multiconfigurational Self-Consistent Field (MCSCF)
Theory.
In the MCSCF approximation, a wave func-
tion Ψ
0−MCSCF
I
(r; R) =
α
c
I
α
ψ
α
(r; R) of the form in
(31.36) is to be determined. In this procedure, both
the molecular orbitals φ(r
j
; R) andc
I
(R) are determined
from the requirement that
E
0−MCSCF
I
=
Ψ
0−MCSCF
I
(r; R)
H
0
Ψ
0−MCSCF
I
(r; R)
r
,
(31.37)
be a minimum. In a popular variant, the state aver-
aged MCSCF (SA-MCSCF) [31.50–53] procedure, the
average energy functional
E
0−SA−MCSCF
=
K
I=2
w
I
Ψ
0−MCSCF
I
(r; R)
H
0
× Ψ
0−MCSCF
I
(r; R)
r
, (31.38)
where the weight vector w = (w
1
,w
2
,... ,w
K
) has
only positive elements, is minimized. This procedure
should be compared with the multireference CI ap-
proach described below in which a predetermined set
of molecular orbitals is used.
The optimum molecular orbitals and CI coeffients
can be written as unitary transformations of an initial set
of such quantities, i. e.,
Ψ
0−MCSCF
I
= exp(γ) exp(∆)Φ
0
I
, (31.39)
where
γ =
i,s
γ
s,i
a
†
s
a
i
,∆=
n,l
∆
l,n
Φ
0
l
Φ
0
n
,
(31.40)
and γ ,∆ are general anti-Hermitian matrices. Since
m
i, j
=−m
j,i
≡ m
ij
for m = γ ,∆, the upper triangle
of m forms a vector
m that enumerates the independent
parameters of m.TheMCSCF or SA-MCSCF equations
can be succinctly formulated by inserting (31.39)into
(31.37)or(31.38), expanding the commutators to second
order and requiring ∂E/∂m
ij
= 0 [31.36]. The result pro-
vides a system of Newton-Raphson equations that can
be solved iteratively for the γ , ∆ [31.52, 53,67,73].
Multireference Configuration Interaction Theory: The
MR-SDCI Method.
In multireference configuration inter-
action theory, the wave function is again of the form
Ψ
0−MRCI
I
(r; R) =
α
c
I
α
(R)ψ
α
(r; R), but now CSFs
involving the large space of virtual orbitals are included.
In this approach the CI coefficients are found for a prede-
termined set of molecular orbitals. The CSF expansions
at the MR-SDCI level become quite large (1–10 mil-
lion CSFs is routine), and even larger expansions are
tractable using specialized methods. The c
I
satisfy the
usual matrix equation
H
0
− E
0
I
c
I
= 0 , (31.41)
where
H
0
αβ
=ψ
α
|H
0
|ψ
β
=
i, j
A
αβ
ij
h
ij
+
i, j,k,l
A
αβ
ijkl
(ij|kl). (31.42)
It is the computationally elegant solution of (31.41)for
largeexpansionsthat is the essence of modern MR-SDCI
methods.
Because of the large dimension of the CSF space, it is
not possible (or even desirable) to find all the solutions
of (31.41). The few lowest eigenstates and eigenval-
ues can be found [31.74] using an iterative direct CI
Part C 31.2
Molecular Structure 31.2 Characterization of Potential Energy Surfaces 475
procedure [31.75] in which a subspace is generated se-
quentially from the residual defined at the kth iteration
by
σ
(k)
µ
=
ν
H
0
µν
− E
(k−1)
δ
µν
c
(k−1)
ν
, (31.43)
where for simplicity of notation, the state index
I is suppressed. The computationally demanding
step in this procedure is the efficient evaluation of
the A
αβ
ijkl
=ψ
α
|
˜
E
ij
˜
E
kl
− δ
jk
˜
E
il
|ψ
β
. Key to the effi-
ciency of this evaluation is the factorization formally
achieved by
ψ
α
|
˜
E
ij
˜
E
kl
|ψ
β
=
m
ψ
α
|
˜
E
ij
|ψ
m
ψ
m
|
˜
E
kl
|ψ
β
.
(31.44)
Using unitary group techniques, this apparently in-
tractable summation can be used to express the A
µν
ijkl
as a simple finite product [31.76].
Contracted CI and Complete Active Space Perturba-
tion Theory (CASPT 2).
The direct approach outlined
above makes treatment of large MR-SDCI expansions
possible. However, as the size of the reference space
grows, the CSF space in the MR-SDCI expansion may
become intractably large, particularly if the full second-
order wave function is used. To avoid this bottleneck,
the reference CSFs may be selected from the active
space, and perturbation theory may be used to select
CSFs involving orbitals in the virtual space [31.42].
The use of selection procedures complicates the im-
plementation of ‘direct’ techniques although recently
progress in selected direct CI procedures has been re-
ported [31.77–79]. Alternatively, new techniques have
been developed that avoid this selection procedure. In
these approaches, the MCSCF wave function itself is
used as the reference wave function for CI or pertubation
theory techniques. The use of a reference wave function
rather than a reference space considerably reduces the
size of the CSF space to be handled. In this approach,
one of the principal computational complications is
that the excited functions are not necessarily mutually
orthogonal. Two computational procedures currently
in wide use, known as contracted CI [31.80–83]and
CASPT2 [31.84,85], are based on this approach.
The CASPT2 method is a computationally efficient
variant of second order perturbation theory in which the
reference wave function is a CAS-MCSCF wave func-
tion and thus may itself contain tens of thousands of
CSFs. In this case the full multireference CI problem
would be intractable owing to the large space of double
excitations. A similar approach is adopted in the con-
tracted CI method, in that the excitations are defined
in terms of a general MCSCF reference wave func-
tion Ψ
0−MRF
I
rather than the reference space as in the
MR-SDCI methods described above.
31.2.3 Electron Correlation:
Density Functional Theory
The approaches in Sect. 31.1.1 and 31.1.2 can be re-
ferred to as wave function based approaches in the
sense that determination of E
0
I
(R) is accompanied by
the determination of the corresponding electronic wave
function Ψ
0
I
(r; R). An alternative approach is known as
density functional theory (DFT) [31.86]. The ultimate
goal of DFT is the determination of total densities and
energies without the determination of wave functions,
as in the Thomas–Fermi approximation. DFT is based
on the Hohenberg–Kohn Theorem [31.87], which states
that the total electronic density can be considered to be
the independent variable in a multi-electron theory (see
also Chapt. 20). Computationally viable approaches ex-
ploit the Kohn and Sham formulation [31.88], which
introduces molecular orbitals as an intermediate device.
The essential features of the Kohn–Sham (KS)the-
ory [31.86] are as follows [31.89]. Assume that the
real N-electron system for a particular arrangement
of the nuclei R has a total electron density ρ(r; R).
Consider a system of N independent noninteracting
electrons subject to a one-body potential V
0
with to-
tal density ρ
0
(r; R) such that ρ(r; R) = ρ
0
(r; R).The
corresponding independent particle orbitals, the Kohn–
Sham orbitals φ
KS
i
(r
j
; R), satisfy a Hartree–Fock-like
equation
−
1
2
∇
2
+ V
0
− ε
i
φ
KS
i
= 0 (31.45)
with
ρ
0
(r; R) =
i
|φ
KS
i
|
2
. (31.46)
The relation between the energy of the ideal system and
that of the true system E
0−DFT
(R) is obtained from the
adiabatic connection formula [31.89].
In order to determine E
0−DFT
(R), functions φ
KS
i
are
required, which in turn means that V
0
, the Kohn–Sham
noninteracting one-body potential, must be determined.
V
0
is written as (31.3)
V
0
= V
e−N
+ V
Coul
+ V
xc
(31.47)
where V
Coul
is the Coulomb interaction corresponding
to the electron density, V
e−N
is the electron-nuclear at-
Part C 31.2
476 Part C Molecules
traction interaction and V
xc
is the exchange-correlation
density. The effect of (31.47) is to isolate from V
0
the straightforward contributions V
Coul
and V
e−N
,and
transfers our ignorance to the remaining portion V
xc
.
Although, by the Hohenberg–Kohn theorem V
xc
must
exist, its determination remains the challenge of modern
density functional theory, which currently uses approx-
imate functional forms. These approximate treatments
of V
xc
are quite useful in practice, and for large systems,
DFT offers a promising alternative to wave function
based methods.
Through (31.45), the KS approach is formally sim-
ilar to SCF theory, although the Kohn–Sham theory is
in principle exact. This formal similarity is exploited in
the evaluation of the derivative E
0−DFT
(R) [31.90].
31.2.4 Weakly Interacting Systems
When attempting to describe weak chemical interac-
tions such as van der Waals or dispersion forces, the
techniques outlined in Sect. 31.2.2 must be modified
somewhat. These modifications arise from the finite-
ness, and hence incompleteness, of the basis (the set χ)
used to describe the molecular orbitals. Assume that
the interaction of two molecules A and B is to be de-
termined. Consider the description of molecule A as
the distance between A and B decreases from infin-
ity. The atom centered basis functions on molecule B
augment those on molecule A, lowering its energy, inde-
pendent of any physical interaction. This computational
artifact serves to overestimatethe interaction energy, and
is known as the basis set superposition error [31.91,92].
In chemically bonded systems where interaction ener-
gies are large, it is of negligible importance. However in
weakly bonded systems for which the interaction ener-
gies may be on the order of 10 to 100 cm
−1
, the basis
set superpostion error can be significant.
The basis set superposition error can be reduced by
the counterpoise correction [31.93,94]. In this approach,
the interaction energy is evaluated directly as
E
0−int
I
(R) = E
0
I
(R) −
E
0−A
I
(R) + E
0−B
I
(R)
,
(31.48)
where E
0−A
I
(R) and E
0−B
I
(R), the energies of A and B
respectively, are evaluated in the full basis.
31.3 Intersurface Interactions: Perturbations
The existence of interstate interactions can lead to ‘unex-
pected’ shifts in spectral lines as well as predissociation
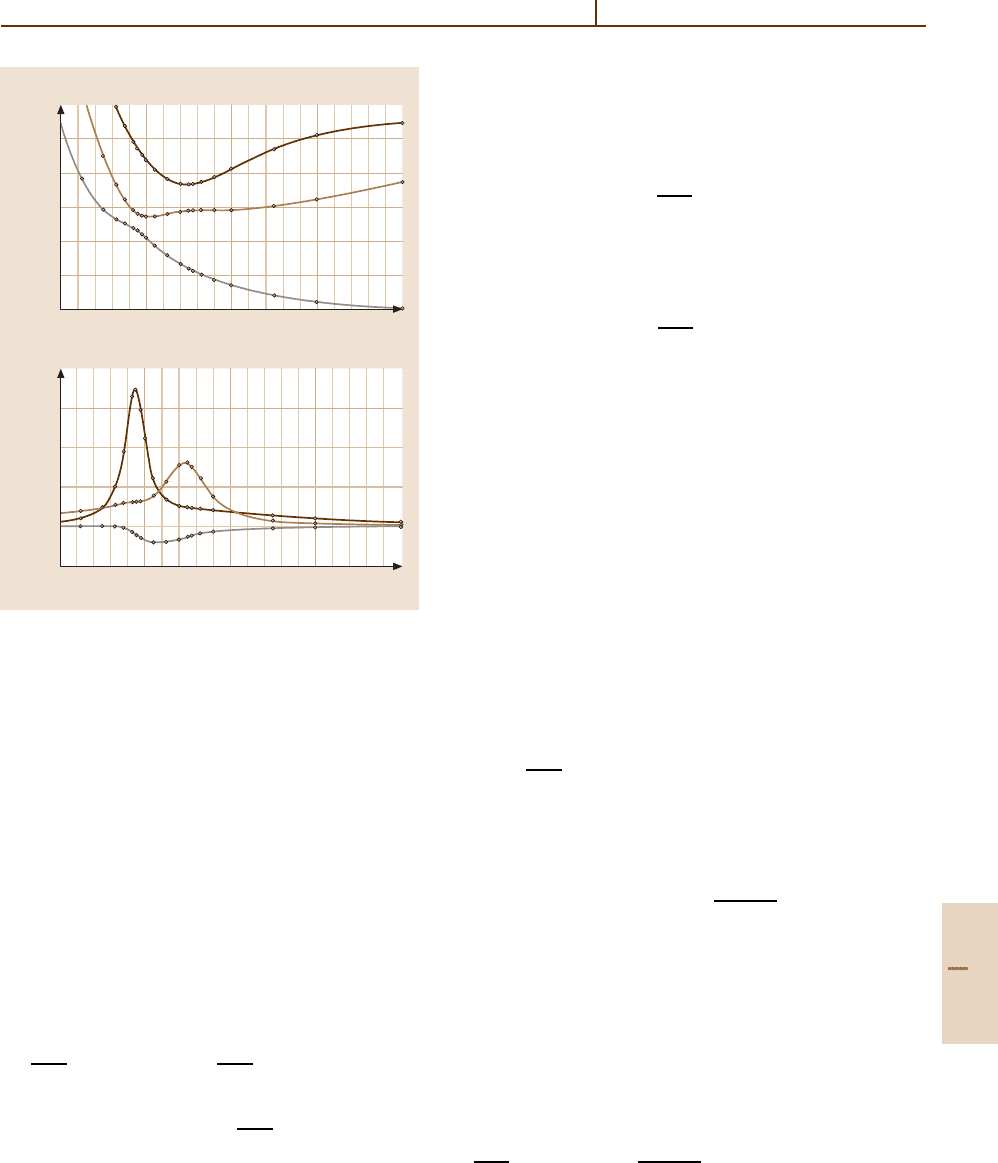
of the states themselves [31.95]. These situations are il-
lustrated in Fig. 31.2a,b which present the 1, 2, 3
3
Π
g
potential energy curves for Al
2
and the corresponding
derivative couplings f
IJ
(R) respectively. The derivative
couplings were evaluated using the method described
in Sect. 31.3.1. In this molecule, derivative couplings
between the 2, 3
3
Π
g
states are responsible for the
perturbations in the vibrational levels of the bound
2, 3
3
Π
g
states. Derivative couplings of these states with
the 1
3
Π
g
state causes predissociation of all levels in
the 2, 3
3
Π
g
manifold [31.96].
Section 31.1 describes two classes of interstate
matrix elements that can lead to these nonadiabatic
phenomena, the derivative coupling matrix elements in
(31.7–31.10)andH
rel
IJ
, which is usually treated for low
Z systems within the Breit–Pauli approximation. An il-
lustration of a nonadiabatic process induced by H
rel
is
provided in Sect. 31.5. A key issue in the treatment of
the electronic structure aspects of these phenomena is
the reliable evaluation of the interstate matrix elements.
The interstate interactions are usually of most interest
in regions of nuclear coordinate space far removed from
the equlibrium nuclear configuration. Thus it is desir-
able to evaluate these interactions using multireference
CI wave functions which (Sect. 31.1) are well suited for
use in these regions of coordinate space. The evaluation
of nonadiabatic interactions, based on SA-MCSCF/CI
wave functions, is discussed below.
31.3.1 Derivative Couplings
From (31.9–31.11), two classes of matrix elements are
required,
˜
k
JI
W
α
W
β
(R) and f
JI
W
α
(R). In fact, techniques
to evaluate both of these exist [31.44]. However, it
is common to approximate
˜
k
JI
W
α
W
β
(R) by
˜
k
JI
W
α
W
β
(R) =
M
∂
∂W
α
Ψ
0
J
(r; R)
Ψ
0
M
(r; R)
r
×
Ψ
0
M
(r; R)
∂
∂W
β
Ψ
0
I
(r; R)
r
=
M
f
MJ
W
α
f
MI
W
β
. (31.49)
with the (in principle infinite) summation over states
truncated to reflect only the states explicitly treated in the
Part C 31.3
Molecular Structure 31.3 Intersurface Interactions: Perturbations 477
40 000
35 000
30 000
25 000
20 000
15 000
10 000
3.5 4.5 5.5 6.5 7.5
4
3
2
1
0
–1
3.5 4.5 5.5 6.5 7.5
E(cm
–1
)
a)
b)
R(Al – Al) (a
0
)
R(Al – Al) (a
0
)
f
IJ
(a
0
–1
)
3
3
Π
g
2
3
Π
g
1
3
Π
g
Al
2
f
12
f
23
f
13
Al
2
Fig. 31.2 (a) Adiabatic potential energy curves for the
1, 2, 3
3
Π
g
states of Al
2
from [31.96]. (b) Derivative coup-
lings F
Ij
(R) for (I, J) = 1, 3
3
Π
g
, 1, 2
3
Π
g
and 2, 3
3
Π
g
from [31.96]
nonadiabatic dynamics. Thus it is sufficient to discuss
the determination of f
JI
W
α
(R).
The use of analytic derivative theory [31.97, 98]
greatly improves the computational efficiency of eval-
uating f
JI
W
α
(R) relative to the earlier divided difference
techniques [31.99]. The key ideas are given below. In this
presentation, the standard, real-valued normalization is
used. Additional contributions owing to the geometric
phase, if required, must be evaluated separately as they
do not follow from the electronic Schrödinger equation
at a single point [31.100].
Differentiation of the Ψ
0
I
(r; R) defined in (31.36)
gives:
∂
∂W
α
Ψ
0
I
(r; R) =
λ
∂
∂W
α
c
I
λ
(R)
ψ
λ
(r; R)
+ c
I
λ
(R)
∂
∂W
α
ψ
λ
(r; R)
.
(31.50)
Thus f
JI
W
α
(R), consists of two terms
f
JI
W
α
(R) =
CI
f
JI
W
α
(R) +
CSF
f
JI
W
α
(R), (31.51)
where the CI contribution is given by
CI
f
JI
W
α
(R) =
λ
c
J
λ
(R)
∂
∂W
α
c
I
λ
(R)
, (31.52)
and the CSF contribution has the form
CSF
f
JI
W
α
(R)
=
λ,µ
c
J
λ
(R)
ψ
λ
(r; R)
∂
∂W
α
ψ
I
µ
(r; R)
r
c
I
µ
(R).
(31.53)
Evaluationof
CSF
f
JI
W
α
(R) is straightforward using analyt-
ical derivative techniques [31.101], so that the remainder
of the discussion focusses on
CI
f
JI
W
α
(R).From(31.52),
it would appear that the derivative of the CI coefficients
∂/∂W
α
c
I
λ
(R) ≡ V
I
W
α
,λ
(R) would be required to evaluate
CI
f
JI
W
α
(R). This is quite costly and is in fact not neces-
sary, since only the projection onto the state Ψ
0
J
(r; R)
is required. Equation (31.52)for
CI
f
JI
W
α
(R) can be recast
in a form similar to that of g
I
(R). This transformation
of (31.52) enables the explicit determination ofV
I
W
α
(R)
to be avoided, and is the key to the efficient use of
analytic gradient techniques in the evaluation of f
JI
(R).
Differentiating (31.41) with respect to W
α
gives
H
0
− E
0
I
(R)
V
I
W
α
(R)
=−
∂
∂W
α
H
0
− E
0
I
(R)
c
I
(R). (31.54)
Taking the inner product of (31.54) with c
J
(R) gives
CI
f
JI
W
α
(R) ≡ c
J
(R)
†
V
I
W
α
(R) (31.55)
= ∆E
0
IJ
(R)
−1
c
J
(R)
†
∂ H
0
(R)
∂W
α
c
I
(R)
(31.56)
≡ ∆E
0
IJ
(R)
−1
h
JI
W
α
(R). (31.57)
Observe that (31.56)and(31.57) are not the Hellmann–
Feynman theorem [31.101, 102](Chapt.51), to which
they bear a formal resemblance, since it is not the
Hamiltonian operator H
0
(r; R) but rather the Hamil-
tonian matrix H
0
(R) that is being differentiated. Since
the energy gradient has the form [31.44]
∂
∂W
α
E
0
I
(R) = c
I
(R)
†
∂ H
0
(R)
∂W
α
c
I
(R), (31.58)
Part C 31.3

478 Part C Molecules
its relation to
CI
f
JI
W
α
(R) is clear. This identification is the
keystepintheevaluationof
CI
f
JI
W
α
(R) using analytic
gradient techniques [31.97,98].
31.3.2 Breit–Pauli Interactions
For light systems, it is possible to introduce relativistic
effects using the Breit–Pauli approximation [31.1], in
which the four component Dirac description of a sin-
gle electron is replaced by a two component (α, β)
description. The H
e
(r; R) becomes [31.1,103]
H
e
(r; R) = H
0
(r; R) + H
rel
(r; R), (31.59)
where, in parallel with (31.4–31.13) for the atomic case,
the relativistic correction H
rel
=
3
k=1
H
k
(r; R) can be
divided into spin-dependent (SD) and spin-independent
(SI) parts, plus an external field interaction term H
ext
.
These are given by
H
1
= H
SD
≡ H
so
+ H
soo
+ H
ss
, (31.60)
H
2
= H
SI
≡ H
mass
+ H
D
+ H
ssc
+ H
oo
,
(31.61)
H
3
= H
ext
, (31.62)
where, in atomic units,
H
so
=
α
2
2
K,i
Z
K
(r
i
− R
K
) × p
i
· s
i
|
r
i
− R
K
|
3
, (31.63)
H
soo
=−
α
2
2
i= j
r
ij
× p
i
· (s
i
+ 2s
j
)
r
ij
3
, (31.64)
H
ss
= α
2
i< j
s
i
· s
j
|r
ij
|
3
−
3(r
ij
· s
i
)(r
ij
· s
j
)
|r
ij
|
5
,
(31.65)
H
mass
=−
α
2
8
i
p
4
i
, (31.66)
H
D
=−
α
2
8
K,i
Z
K
∇
2
i
|
r
i
− R
K
|
−1
−
i= j
∇
2
i
r
ij
−1
,
(31.67)
H
ssc
=−
8πα
2
3
i< j
(s
i
· s
j
)δ(r
ij
), (31.68)
H
oo
=−
α
2
4
j=i
p
i
· p
j
|r
ij
|
−
r
ij
·
r
ij
· p
j
p
i
|r
ij
|
3
,
(31.69)
H
ext
=
α
2
2
i
E(r
i
) × p
i
· s
i
+
iα
2
4
i
E(r
i
) · p
i
+ 2µ
i
H (r
i
) · s
i
, (31.70)
E(r) and H (r) are electric and magnetic fields,
and µ = e
/(2m
e
) is the Bohr magneton. The phys-
ical significance of these terms is discussed is
Sect. 21.1. One of the most important consequences
of these relativistic effects is that total electron
spin is no longer a good quantum number as it
is in H
0
(r; R).ThetermH
SD
couple states corre-
sponding to distinct eigenvalues of S
2
and lead to
the nonadiabatic effects that are the subject of this
section.
The Breit–Pauli approximation is most useful for
light atoms, but the approximation breaks down when
Z becomes large [31.104–106]. One of the principal
effects omitted in a treatment which includes only
H
SD
is the relativistic contraction of the molecular
orbitals [31.107] due to the mass-velocity operator
(H
mass
), an effect whose importance increases with Z.
Several approaches exist which attempt to correct this
situation while retaining the spirit and simplifications
of the Breit–Pauli approximation. The first of these is
the relativistic effective core potential (ECP) approxi-
mation [31.108–111]. In this approach, the results of
an atomic Dirac–Fock calculation [31.112]areusedto
replace innermost or core electrons of a given atom
with (i) an effective one electron potential that mod-
ifies the electron-nuclear attraction term in H
0
and
(ii) an effective one electron spin-orbit operator, so
that
H
e
(r; R) → H
e−ECP
(r; R)
= H
0−ECP
(r; R) + H
so−ECP
(r; R).
(31.71)
The formal similarity between H
e−ECP
and H
e
(r; R)
results in a similar phenomenological interpretation of
relativisticallyinduced nonadiabatic processes. Applica-
tions of this approach have been reviewed [31.107,113].
A second approach includes all electrons explic-
itly, and uses H
SD
as defined above. The relativistic
contraction of the core electrons is included by
using a variational one-component spin-free approxima-
tion [31.114, 115] to the no-pair Hamiltonian [31.116]
at the orbital optimization stage. The variational na-
ture of the approximation provides advantages over
the use of the H
mass
term. Applications of this ap-
proach to the spectra of CuH and NiH have been
reported [31.117,118].
Part C 31.3
Molecular Structure 31.3 Intersurface Interactions: Perturbations 479
In order to evaluate H
SD
IJ
(R), it is necessary to
specify the molecular orbitals to be used to con-
struct the Ψ
0
I
(r; R). The choice of molecular orbitals
is dictated by the following considerations. Matrix el-
ements of H
SD
between different states are required.
The molecular orbitals appropriate for one state may
not be appropriate for the description of the sec-
ond state. Two approaches are available to handle
this situation. In one approach, distinct sets of (mu-
tually nonorthogonal) molecular orbitals are used to
describe each state [31.119]. This permits a more com-
pact description of the spaces in question. However
in this case one is required to evaluate the ma-
trix element of a two electron operator H
soo
and/or
H
ss
in a nonorthogonal molecular orbital basis, an
imposing computational task. This significantly lim-
its the size of the CSF space which is tractable.
The alternative approach is to use a common or-
thonormal basis balanced between the two spaces in
question, and to use larger CSF spaces [31.120–122].
The use of a common orthonormal basis decreases
significantly the computational effort required to eval-
uate the matrix elements. A symbolic matrix element
method [31.123] has been applied to H
SD
, as described
in the review [31.124].
31.3.3 Surfaces of Intersection
The preceding subsections have considered what must
be calculated in order to characterize an electron-
ically nonadiabatic process. As noted in Sect. 31.1,
it is also necessary to consider where in nuclear
coordinate space electronic nonadiabaticity is impor-
tant. Nonadiabatic processes are important in regions
of close approach of the potential energy surfaces
with regions of surface intersections being of pre-
eminent interest. Until recently, these surfaces of
intersection were determined by indirect methods,
i. e., the potential energy surfaces were characterized,
and then the surface of intersection was determined.
This made the determination of these surfaces of
intersection a computationally daunting task. How-
ever, computational advances have made it possible
to determine these surfaces of intersection directly,
i. e., without prior determination of the individual
potential energy surfaces. This point is discussed
next.
A point on the surface of conical intersec-
tion of two states of the same symmetry, subject
to a set of geometric equality constraints of the
form C
i
(R) = 0, i = 1,... ,m, is determined from the
Newton–Raphson equations [31.125]
−
Q
IJ
(R, ξ, λ) g
IJ
(R) h
IJ
(R) k(R)
g
IJ
(R)
†
000
h
IJ
(R)
†
000
k(R)
†
0
†
0
†
0
δR
δξ
1
δξ
2
δλ
=
g
I
(R) + ξ
1
g
IJ
(R) + ξ
2
h
IJ
(R) +
m
i=1
λ
i
k
i
(R)
∆E
IJ
(R)
0
C(R)
(31.72)
where δR= R
− R,δλ = λ
− λ,δξ = ξ
− ξ, g
IJ
α
(R) ≡
∂∆ E
IJ
(R)/∂R
α
, k
i
α
(R) ≡ ∂C
i
(R)/∂R
α
, h
IJ
α
(R) ≡ c
I †
(R)H
0
(R)/∂R
α
c
J
(R) ,ξ and λ are Lagrange multipli-
ers, and Q
IJ
(R, ξ, λ) is a matrix of second derivatives
[31.125]. For two states of different symmetry, the ana-
logue of (31.72) is used with the terms related to ξ
2
omitted [31.126]. The excellent performance of this
algorithm has been documented [31.125,127].
Equations (31.72) can be motivated as follows. R
c
is
sought so that E
0
I
(R) is minimized subject to the con-
straints E
0
I
(R) = E
0
J
(R) andC(R) = 0. The key is to
impose the first of these constraints, noting that at each
step in the Newton–Raphson procedure,c
I
(R) andc
J
(R)
are eigenvectors. Equation (31.72) are the Newton-
Raphson equations corresponding to the Lagrangian
function [31.128]
L
IJ
(R, ξ, λ) = E
I
(R) + ξ
1
∆E
IJ
(R)
+ ξ
2
H
IJ
(R) +
M
k=1
λ
k
C
k
(R),
(31.73)
provided that the gradient of H
0
IJ
(R) is interpreted as
a change in H
0
(R) within the subspace spanned by
c
I
(R) and c
J
(R). This can be derived from quaside-
generate perturbation theory and understood as follows.
Assume for convenience that m = 0, i. e., there are no
geometrical constraints.
Consider an R for which (31.72) is not satisfied.
The 2 × 2 matrix H(R) with matrix elements H
KL
(R),
K, L ∈{I, J} becomes at R+ δR
H
IJ
(R+ δR)
= c
I †
(R)
%
H(R) +
α
∂ H(R)
∂R
α
δR
α
&
c
J
(R),
(31.74)
Part C 31.3

480 Part C Molecules
H(R) is diagonal but nondegenerate at R,i.e.,
H(R) =
%
E
0
I
(R) − E
0
J
(R) 0
00
&
.
(31.75)
From (31.74)atR
c
= R+ δR, H(R
c
) becomes, to first
order, ignoring an irrelevant uniform shift of the diagonal
elements,
H(R
c
) =
%
∆E
IJ
(R) + g
IJ †
· δRh
IJ †
· δR
h
IJ †
· δR 0
&
.
(31.76)
Thus (31.72) is seen to be the requirement that H(R
c
)
has degenerate eigenvalues, with eigenvectors c
I
(R) and
c
J
(R).When(31.72) have been solved, H
IJ
(R
c
) is di-
agonal and degenerate, E
I
(R
c
) has been minimized, and
g
I
(R
c
) = 0, except along directions contained in the two
dimensional subspace spanned by g
IJ
(R
c
) and h
IJ
(R
c
).
From (31.76), it is these two directions which lift the
degeneracy ofH(R
c
). Equation (31.72) is also relevant
to the reaction path in a nonadiabatic process as dis-
cussed in Sect. 31.4. A discussion of these points from
an alternative perspective has been presented by Radazos
et al. [31.129].
A solution to (31.72) is referred to as a conic-
al intersection, although rigorously degenerate states
cannot be obtained from a numerical procedure.
If required, the existence of a conical intersec-
tion can be rigorously established by showing that
the adiabatic wave functions undergo a change of
sign when transported around a closed loop con-
taining R
c
in the plane defined by g
IJ
(R
c
) and
h
IJ
(R
c
). This is the geometric or Berry phase crite-
rion [31.130, 131]. It has been explicitly demonstrated
for a conical intersection in O
3
by Ruedenberg and
coworkers [31.132].
31.4 Nuclear Motion
31.4.1 General Considerations
The determination of the rovibrational spectrum of
polyatomic systems from first principles is a problem
of primary importance since it allows the deter-
mination of molecular forces and structure from
spectral data. In the adiabiatic case, the solution
of (31.6)
T
nuc
(R) + E
0
I
(Q) − E
K
β
K
(R) = 0 (31.77)
is sought, where Q denotes a set of internal nuclear coor-
dinates. The reliability of the solution of (31.77) reflects
the accuracy of the Born–Oppenheimer potential energy
surface E
0
I
(Q) appearing in that equation. The meth-
ods for determining E
0
I
(Q) were discussed in Sect. 31.2.
Conversely, the determination of the E
K
from spectro-
scopic measurements can be used to infer information
concerning E
0
I
(Q). In either case, the accurate solution
of (31.77) is requisite and this section is concerned with
its solution.
The operator in (31.77) has a continuous spectrum
since T
nuc
includes translations of the nuclear center
of mass (cm). An operator with a discrete spectrum
is obtained by replacing the Hamiltonian in (31.77)
with one in which the translation of the nuclear c.m.
has been eliminated by transforming to the c.m. frame.
In the center of mass frame, (31.77) has the general
form [31.133]
T
vr
(Ω, Q) + T
vib
(Q) + E
0
I
(Q) − E
K
β( , Q) = 0
(31.78)
where Ω are three rotational coordinates and T
vr
and
T
vib
are the rotational and vibrational kinetic energy op-
erators. In T
vr
(Ω, Q) all the complexity associated with
the coupling of nuclear and electronic angular momen-
tum is buried. The determination of the appropriate form
for (31.78) is by no means straightforward [31.134,135],
and treatment of the effects of multiple angular mo-
mentum is a complex problem in angular momentum
algebra.
In diatomic systems, the vibrational problem
involves only one internal coordinate and is straightfor-
ward. Angular momentum coupling is usually treated
using Hund’s case (a), (b), etc., or an intermediate
case approach [31.95] with Van Vleck’s reversed an-
gular momentum commutation relations being helpful
in analyzing the coupled angular momentum prob-
lem [31.134].
Polyatomic systems introduce new complications,
since in addition to the increased dimensionality of
the vibrational problem, internal and rotational coor-
dinates interconvert for colinear arrangements of the
nuclei; this is particularly relevant in triatomic systems.
In addition, since the C
∞v
point group has doubly degen-
erate representations at collinear geometries, electronic
Part C 31.4

Molecular Structure 31.4 Nuclear Motion 481
state degeneracies may arise (the Renner–Teller ef-
fect) [31.32,136,137], further complicating the analysis.
Techniques associated with the description of triatomic
systems involving coupled angular momentum and elec-
tronic degeneracy are illustrated in Sect. 31.4.3.
More generally, (31.78) can be rewritten as
T
vr
(Ω, Q
e
) + T
vib
(Q) + E
sep
I
(Q) − E
K
β(R)
=−∆T
vr
(Ω, Q)β(R) − ∆E
0
I
(Q), (31.79)
where
∆T
vr
(Ω, Q) ≡ T
vr
(Ω, Q) − T
vr
(Ω, Q
e
), (31.80)
∆E
0
I
(Q) ≡ E
0
I
(Q) − E
sep
I
(Q), (31.81)
Q
e
is an equilibrium structure, and E
sep
I
(Q) is a separa-
ble function of the normal coordinates. In this case, the
solution to the left-hand side of (31.79) can be factor-
ized into a rotational part and a vibrational part [31.138]
according to
ψ
J
l
=
K
k
J
K
D
J
K0
(θ, χ)
i
α
l
i
(Q
i
) (31.82)
where D
J
K0
is a symmetric top wave function [31.139],
and α
j
(Q
i
) is a pure vibrational wave function, e.g.,
harmonic oscillator function, for the ith internal normal
coordinate. It is in this approximation that the notion
of a molecule rotating and vibrating about a fixed mo-
lecular structure is achieved. In this case, E
K
is just
the sum of the pure rotational and vibrational energies.
These rigid rotator-vibrator solutions then form the basis
for the inclusion of the effects of the right-hand side us-
ing, for example, perturbation theory [31.140]. They can
also be used as a basis for a nonperturbative treatment
(Sect. 31.4.2).
The classic treatments of (31.77) [31.141–144]have
been periodically revisited [31.134, 145]. In these in-
vestigations, the contributions of electronic angular
momentum to this Hamiltonian were frequently sup-
pressed [31.144] so that these treatments are appropriate
to totally symmetric electronic states. Van Vleck [31.134]
has shown how the effects of electronic angular momen-
tum can be incorporated into these treatments.
For nonlinear polyatomic systems, the Hamiltonian
in (31.78) can be transformed to the Eckart or body
fixed frame in which the reference axis is a body
fixed axis oriented along the principal moments of in-
ertia [31.146], although other choices of the body fixed
axes are possible [31.147]. The principal moments of
inertia represent the eigenvectors of the inertial tensor
matrix I
ij
, i, j = x, y, z, where [31.144]
I
xx
=
i
M
i
(R
e
i
· R
e
i
− X
e
i
X
e
i
), (31.83)
I
xy
=−
i
M
i
(X
e
i
Y
e
i
), (31.84)
and cyclic permutations, and R
e
denotes an equilibrium
structure. The Hamiltonian determined by this proce-
dure is referred to as the Watson Hamiltonian [31.145],
and is widely used in discussing the rovibrational
spectrum of nonlinear polyatomic molecules in singlet
electronic states. Treatments of the Watson Hamilto-
nian are mentioned in Sect. 31.4.2. For linear molecules,
an alternative treatment is required since there is one
feweroverall rotational coordinate and one more internal
coordinate [31.138].
31.4.2 Rotational-Vibrational Structure
Various approaches to the solution of (31.79) exist. The
principal issues, which are interrelated, are (i) the range
of nuclear configurations over which E
0
I
(Q) is known,
(ii) the coordinate system used to express the internal
coordinates, and (iii) particularly in larger systems, the
number of modes or internal coordinates retained in the
calculations (Sect. 31.4.3). With regard to point (i), two
approaches are currently in use. The force field method
uses a power series expansion of E
0
I
(Q) about E
0
I
(Q
e
),
that is (using the Einstein summation convention)
E
0
I
(Q) = E
0
I
(Q
e
) +
1
2
∂
2
E
0
I
(Q
e
)
∂Q
i
∂Q
j
∆Q
i
∆Q
j
+
1
6
∂
3
E
0
I
(Q
e
)
∂Q
i
∂Q
j
∂Q
k
∆Q
i
∆Q
j
∆Q
k
(31.85)
together with perturbation theory to determine spec-
troscopic constants. In this approach, the force fields
[the partial derivatives in (31.85) are usually eval-
uated directly with the aid of analytic gradient
techniques (Sect. 31.2). Since the expansion of E
0
I
(Q)
is truncated, the results are not independent of the
coordinate system used. For example, significant differ-
ences in the description of Fermi-resonance parameters
[31.148, 149] in rectilinear and curvilinear coordi-
nates [31.150] have been reported.
Alternatively, E
0
I
(Q) can be represented by a grid
of points around Q
e
. In the most reliable calcula-
tions reported to date, E
0
I
(Q) is determined using
the coupled cluster techniques discussed in Sect. 31.2.
Then (31.79) can be solved in a basis analogous to
Part C 31.4

482 Part C Molecules
ψ
J
l
in (31.82). This approach is frequently referred
to as the vibrational CI problem [31.151]. The relia-
bility of the results depends to a considerable extent
on the basis functions and coordinate system used to
describe the problem. Considerable success has been
reported for a technique in which the Watson Hamil-
tonian and harmonic oscillator functions are used to
solve (31.79) [31.151,152].
31.4.3 Coupling of Electronic
and Rotational Angular Momentum
in Weakly Interacting
An understanding of the molecular structure of the
weakly bound compounds of noble gas atoms and
diatomic molecules provides important insights into
the nature of chemical bonding. The inference of
structural data from spectroscopic observations is an
important aspect of this problem. In this subsec-
tion, a theoretical framework for understanding the
spectroscopy of these systems is outlined as an il-
lustration of the methods used to treat coupling of
electronic and nuclear angular momentum in weakly
interacting triatomic molecular systems. Detailed dis-
cussion of this class of problems can be found
in [31.153–155].
As an example, consider the rovibronic structure
of a noble gas-diatom complex Rg-AB, in either its
1
Σ
+
or
1
Π states (which may be closely spaced). Note
that degeneracy of the
1
Π state will only persist for
collinear geometries of the triatom system. The rovi-
brational wave functions can be expanded in a product
basis of functions describing (i) the rovibronic struc-
ture of AB and (ii) the relative motion (vibrational and
end-over-end rotation) of Rg and AB. Since total angu-
lar momentum J and its space fixed projections M are
good quantum numbers, the rovibronic wave function
can be expanded as
Ψ
JM
=
1
R
v jΩlε
C
JM
jΩlεv
(R)
×|ψ
Rg
|v
Ij
|IjΩε, l; JM , (31.86)
where ψ
Rg
is the ground state wave function for the
noble gas atom, l denotes the angular momentum asso-
ciated with the end-over-end motion, and v, j,εand I
denote respectively the vibrational, angular momentum
(electronic + rotational), e/f symmetry index and state
label of the electronic state of the AB molecule in
Hund’s case (a) basis. The angular momentum coup-
ling algebra noted above is reflected in the definition of
|IjΩε, l; JM,whichis
|IjΩε, l; JM
=
m
j
m
l
jm
j
lm
l
|JM Y
lm
l
(θ, φ)ψ
Ijm
j
Ωε
(β, α) ,
(31.87)
where θ, φ and β, α are the polar and azimuthal an-
gles for the line connecting the noble gas atom to
the center of mass of the diatom and the diatom axis,
respectively, and ···|··· is a Clebsch–Gordan coef-
ficient [31.156](Chapt.2). The ψ
Ijm
j
Ωε
are defined in
turn by [31.95]
ψ
Ijm
j
Ωε
=
2J + 1
8π
1/2
×
D
j
m
j
,Ω
(α, β, 0)
∗
|I
2S+1
Λ
+
(Σ )
+ εD
j
m
j
,−Ω
(α, β, 0)
∗
|I
2S+1
Λ
−
(−Σ)
(31.88)
or
ψ
Ijm
j
Ωε
=
2J + 1
4π
1/2
× D
j
m
j
,0
(α, β, 0)
∗
|I
2S+1
Λ(Σ )
(31.89)
when Λ = Σ = 0, where the electronic state has term
symbol I
2S+1
Λ with Ω = Λ + Σ.
The C
JM
(R) satisfy the usual close coupled equa-
tions [31.157,158] Sect. 47.1.1
−1
2µ
d
2
dR
2
+
l(l+ 1)
2µR
2
I−
k
2
2µ
+ V(r)
C
JM
(r)
= 0 ,
(31.90)
where l(l+ 1) and k
2
/2µ designate the diagonal matri-
ces of orbital angular momentum and asymptotic net
scattering energy of individual channels.V(R) repre-
sents the matrix elements of H
e
(r;R) in the vibronic
basis defined in (31.86–31.89). It is built from terms of
the form
Y
lm
D
j ∗
m
j
Ω
v
Ij
ψ
Rg
I
2S+1
Λ
H
e
(r; R)
×
ψ
Rg
I
2S
+1
Λ
r
v
I
j
Y
l
m
D
j
∗
m
j
Ω
≡
Y
lm
D
j ∗
m
j
Ω
v
Ij
H
e
II
(R,
˜
β,r)
Y
l
m
D
j
∗
m
j
Ω
v
I
j
.
(31.91)
The angle
˜
β is the polar angle of the diatom with respect
to the atom-diatom axis. The angular integrations on the
Part C 31.4