Drake G.W.F. (editor) Handbook of Atomic, Molecular, and Optical Physics
Подождите немного. Документ загружается.


316 Part B Atoms
plex for the 3s
2
3p
23
P ground state of Si, is a neon-like
core, 1s
2
2s
2
2p
6
, coupled to the {N = 4,π = even,
3
P}
CAS of the active set {3s, 3p, 3d}.IftheAS is extended
to {3s, 3p, 3d, 4s, 4p, 4d, 4f}, then all single, double,
triple, quadruple (SDTQ) replacements from the outer
four electrons are generated. Of course, the number of
CSF increase rapidly, both with the size of the active
set and with the number of electrons N defining the
CAS. For this reason, other models may be used, such as
multi-reference singles and doubles (MR–SD) modeled
on the results of Z-dependent perturbation theory. These
multi-reference functions may be extended to include all
important contributors to a wave function. Calculations
in which correlation is restricted to a few outer electrons
is called valence correlation.
A consequence of a CAS expansion is that the
MCHF problem is over determined: a rotation of ra-
dial functions of the same symmetry merely transforms
the CSFs and the expansion coefficients. The wave func-
tion and energy do not change. When similar situations
occur in Hartree–Fock theory, Koopmans’ theorem re-
quires that the lagrange multiplier associated with the
rotation be set to zero. In MCHF calculations, the de-
gree of freedom is usually used to eliminate a CSF or,
more precisely, to determine that solution for which
a specific CSF has a zero expansion coefficient. A gener-
alized Brillouin’s Theorem (GBT) then holds. In the case
of the helium ground state (or any ns
2
pair function),
applying GBT leads to the natural orbital expansion
of the form {1s
2
, 2s
2
, 3s
2
,... ,2p
2
, 3p
2
,... ,3d
2
,...}
in which all CSF differ by two electrons. For other
symmetries, such as 1s2p
3
P, a reduced form [21.8]
can also be defined in which all CSF differ by two
electrons, but now involves different sets of orthonor-
mal radial functions for the different partial waves
as in {1s2p
1
, 2s3p
1
, 3s4p
1
,... ,2p
2
3d
1
, 3p
2
4d
1
,...}.
Such expansions yield the fastest rate of convergence,
but are difficult to apply in complex systems. For a his-
tory of Brillouin’s theorem and its use in solving multi-
configuration self-consistent field problems see [21.9].
21.5.4 Excited States
For ground states or atomic states lowest in their
symmetry, the variational procedure is a minimization
procedure, and consequently any approximate energy is
an upper bound. For all others the energies are stationary.
Such calculations may be difficult. One of the most dif-
ficult has been the HF calculation for 1s2s
1
S and, once
obtained is disappointing since the energy is too low. Ex-
cited state calculations become minimization problems
through the use of the Hylleraas–Undheim–MacDonald
theorem (Sect. 11.3.1). Consider a CAS calculation
over the active set {1s, 2s} for which the CSFsare
{1s
2
, 1s2s, 2s
2
}. In determining orbitals, we have a de-
gree of freedom so one CSF may be removed. Selecting
2s
2
has the consequence that the eigenvalue for the 1s2s
state to be determined is now the second eigenvalue, and
hence an upper bound to the exact.
21.5.5 Autoionizing States
The MCHF variational method may be applied to core
excited states imbedded in the continuum, provided that
certain CSFs with filled shells are omitted. An example
is 1s2s2p
2
P of Lithium. An MCHF calculation omit-
ting 1s
2
np configuration states represents the localized
charge distribution of a state in the continuum. How-
ever, the resulting energy is not guaranteed to be an
upper bound to the exact energy.
The saddle-point variational method, as described in
Chapt. 25 is a minimax method for such states.
21.6 Configuration Interaction Methods
Configuration interaction (CI) methods differ from vari-
ational methods like MCHF in that the radial functions
are assumed to be fixed, and hence known in advance.
There are several situations where these methods can be
used effectively:
MCHF with Breit–Pauli
Since the Breit–Pauli operators are valid only as
first order perturbations, variational calculations for
the Breit–Pauli Hamiltonian are not justified. Instead,
MCHF calculations are performed to provide a basis for
LSJ wave functions of (21.14). The expansion coef-
ficients are obtained as a CI calculation. Usually such
expansions are a concatenation of the expansions of all
the LS terms of a configuration and possibly some close
lying configurations. Most Breit–Pauli codes require
a single orthonormal basis. In order to have an orbital
basis that simultaneously describes the correlation of
all these LS terms from a systematic procedure, the
MCHF derivation described earlier has been extended
to derive systems of coupled equations for linear combi-
nations of energy functionals, one for each LS term and
Part B 21.6
Atomic Structure: Multiconfiguration Hartree–Fock Theories 21.6 Configuration Interaction Methods 317
eigenstate. With this extension, it has been possible to
perform “spectrum” calculations in which all LSJ lev-
els up to a certain point in the spectrum are included.
This requires a balanced correlation approach so that
the energy differences with respect to the ground state is
in good agreement with the observed excitation energy.
Once wave functions have been obtained for each level,
transition probabilities can be computed. With all E1
transitions between these levels and some E2/M1 tran-
sitions, the lifetimes of levels can be obtained [21.10].
Full-Core Methods
The full-core plus correlation method (FCPC) [21.16]
is a configuration interaction method. It is mostly used
for an atomic system in which a well defined “core”
is present. For example, in a system such as the three-
elec tron 1s
2
nl or the four-electron 1s
2
nln
l
, the system
has a 1s1s core. In this case, the wave function for an
N-electron system is written as
Ψ(1, 2, 3,... ,N ) =
Aφ
1s1s
(1, 2)
i
C
i
Φ
i
(3, 4,... ,N )
+A
j
D
j
ψ
j
(1, 2, 3,... ,N ),
where φ
1s1s
(1, 2), the wave function of the core, is used
as a single term, and A denotes antisymmetrization. The
relaxation of the core and other correlation effects are
accounted for by the last term in this equation. The cor-
relation effect of the 1s1s core is, in general, very strong
and it is difficult to fully account for this effect in a con-
ventional N-electron wave function. If inaccurate results
are obtained from such a wave function, it may be diffi-
cult to distinguish between errors coming from the core
part or from the other parts. In the FCPC, the correlation
effect in φ
1s1s
(1, 2) can be precalculated to a desired ac-
curacy such that ψ
j
no longer contains the contribution
from the unperturbed core. In physical processes such as
ionization, optical transitions and others, the 1s1s core
wave function in the final state does not change much
from that of the initial state. Using the same φ
1s1s
(1, 2)
for the core may minimize possible errors due to the in-
accuracy of the core wave function. The application of
this method is not limited to systems with 1s1s cores.
For example, for the lithium-like 1s2snp
4
P
o
,forn ≥4,
the 1s2s
3
S can also be considered as an appropriate
“core”.
From a computational view point, the FCPC wave
function has the advantage that it reduces the matrix size
of the secular equation substantially. This drastically
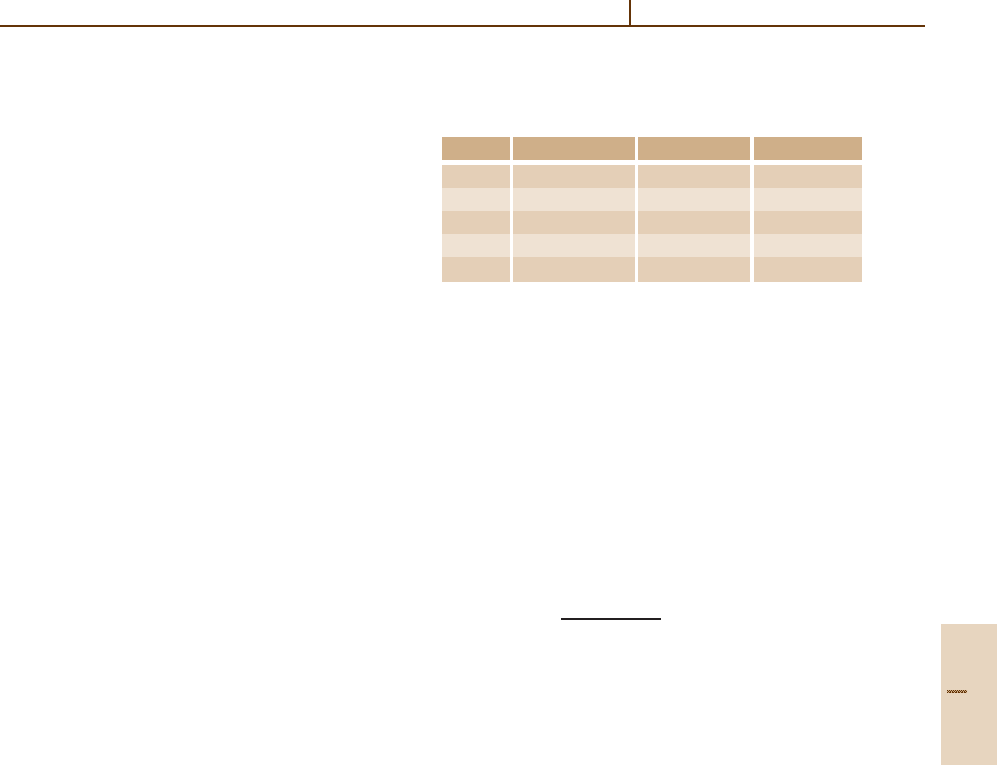
Table 21.3 Comparison of theoretical and experimental en-
ergies for Be 1s
2
2s
21
S in hartrees. All theoretical values
include some form of extrapolation
Ref. Method −E
NR
−E
rel
[21.11] FEM MCHF 14.667 37 14.669 67
[21.12] MCHF 14.667 315
[21.13] Full-core CI 14.667 3492 14.669 6774
[21.14] Semi-empirical 14.667 353
[21.15] Experiment 14.6696759
reduces the memory and CPU time needed on a com-
puter. This method has been quite successful in getting
accurate results for four-electron systems as shown in
Table 21.3. See [21.13] for more details.
Slater Type Orbitals
The essential characteristic of a radial function can be
well represented by the expansion
P
nl
(r) =
j
c
jnl
φ
jnl
(r),
where
φ
jnl
(r) =
(2ζ
jnl
)
2I
jnl
+1
(2I
jnl
)
1/2
r
I
jnl
exp(−ζ
jnl
r)
is a Slater type orbital (STO). Optimized sets of pa-
rameters have been tabulated for many Hartree–Fock
wave functions and these may be used to represent the
core [21.17]. Others may be added and selected orbitals
exponent optimized (only the exponent is varied) so as
to augment the basis. This method has been used effec-
tively by A. Hibbert as implemented in the Configuration
Interaction Version 3 program [21.18].
Spline Basis
The analytic basis methods described in the previous
section have some similarities with MCHF in that linear
combinations of STOs first represent orbitals which then
define the CSFs. These bases result in extensive cancel-
lation as shown by Hansen et al. [21.19]. An expansion
in B-spline basis functions, B
i,k
(r), which are a basis for
a piecewise polynomial subspace (see Chapt. 8), provide
a more flexible basis with very little cancellation in this
representation.
By solving a radial equation with a well chosen po-
tential using the spline Galerkin method that leads to
a matrix eigenvalue problem, a complete set of orbitals
for a piecewise polynomial space can be obtained. The
resulting orthogonal orbital basis may then be used in
Part B 21.6
318 Part B Atoms
CI calculations. Such methods have been reviewed by
Hansen et al. [21.19]. Methods that deal directly with the
primitive B-spline basis have been applied to the study
of Rydberg series [21.20], though orthogonality to tar-
get orbitals was still required. More recently, by using
non-orthogonal theory, an Rmatrix-CI method has been
developed that leads to a generalized eigenvalue prob-
lem. As in the close-coupling approximation, the wave
function is expressed in terms of one or more “targets,”
each coupled to a Rydberg orbital, and an arbitrary num-
ber of pseudo-states. Each Rydberg orbital is expressed
as a linear combination of B-splines but there is no re-
quirement of orthogonality of the Rydberg orbital to the
orbitals defining the target function [21.21]
21.7 Atomic Properties
The discussion so far has concentrated entirely on deter-
mining accurate wavefunctions based on expressions for
the energy. Energy levels are a by-product of such cal-
culations, but once a wave function is known a number
of atomic properties can be evaluated.
21.7.1 Isotope Effects
The isotope shift observed in atomic transitions can be
separated into a mass shift and a field shift. The mass
shift is due to differences in the nuclear mass of the
isotopes, and is the dominant effect for light atoms. The
volume shift arises from the finite volume of the nuclear
charge distribution, and is important for heavy atoms.
From a physical point of view the field shift is the more
interesting, since it yields information about differences
in the nuclear charge distribution between the isotopes.
The isotope shift in a transition is given as the dif-
ference between the shift for the upper and lower level.
The individual shifts are often large, but cancel, and
therefore it is necessary to calculate them very accu-
rately in order to get a reliable value for the difference.
The energyshifts are evaluated in first-order perturbation
theory with wave functions obtained from the zero-order
Schrödinger Hamiltonian, H
0
.
Mass Shift
Up to this point, the nuclear mass has been taken to
be infinite. If instead it has a finite value M,anN-
electron atom turns into an (N+1)-particle dynamical
system. A transformation to center of mass R plus rel-
ative coordinates r
i
= r
i
−r
nuc
yields the transformed
Hamiltonian [21.22]
H =
P
2
2M
tot
+
N
i=1
p
2
i
2µ
+
N
i< j
p
i
· p
j
M
+V
r
i
,
(21.55)
where M
tot
= M + Nm is the total mass and
µ = Mm/(M +m) is the reduced mass. The first term is
the kinetic energy of the center of mass, which can be ne-
glected if Ris an ignorable coordinate. For the remaining
terms, introduce the scaled distances ρ
i
=r
i
/a
µ
,where
a
µ
= (m/µ)a
0
is the scaled Bohr radius. If the potential
is entirely Coulombic, then H assumes the form
H =−
1
2
N
i=1
∇
2
ρ
i
−
µ
M
N
i< j
∇
ρ
i
·∇
ρ
j
+V(ρ
i
)
(21.56)
in units of e
2
/a
µ
. This can be written in the form H =
H
0
+ H
MP
,whereH
0
includes the first and last terms
of (21.56), and
H
MP
=−
µ
M
N
i< j
∇
ρ
i
·∇
ρ
j
e
2
/a
µ
(21.57)
is the additional mass polarization term.
Equation (21.56) gives rise to two kinds of mass
shifts. First, since H is identical to the infinite mass
Hamiltonian, all its eigenvalues E
0
are multiplied by
a
0
/a
µ
= µ/m, resulting in the normal mass shift (nms)
∆E
nms
=−(µ/M)E
0
. (21.58)
Second, if H
MP
is treated as a first order perturbation,
then the resulting specific mass shift (sms) is
∆E
sms
=Ψ
0
| H
MP
| Ψ
0
(e
2
/a
µ
) (21.59)
where Ψ
0
is an eigenvector of H
0
. The specific mass
shift parameter S is defined by
S =
Ψ
0
|−
N
i< j
∇
ρ
i
·∇
ρ
j
| Ψ
0
.
(21.60)
Field Shift
The field shift of an atomic energy level is due to the
extended nuclear charge distribution. The field inside
the nucleus deviates from the Coulomb field of a point
Part B 21.7
Atomic Structure: Multiconfiguration Hartree–Fock Theories 21.7 Atomic Properties 319
charge, and this is reflected in the calculated levels. For
light atoms, the resulting energy correction to the level
E
0
is expressed in terms of the nonrelativistic electron
probability at the origin, |Ψ(0)|
2
:
E
fs
=
2π
3
Z
r
2
N
|Ψ(0)|
2
, (21.61)
where
r
2
N
is the mean square radius of the nucleus. For
heavier atoms (Z > 10) it becomes necessary to include
a relativistic correction factor. Numerical values of this
correction factor can be found in [21.23].
Table 21.4 shows the convergence of an MCHF cal-
culation for the specific mass shift parameter and the
electron density at the nucleus of the ground state of
Boron II as the active set increases. The calculated
11
B−
10
B isotope shift is 13.3 mÅ with an estimated
uncertainty of 1%. The size of the isotope shift is sim-
ilar to the limit of resolution of the Goddard High
Resolution Spectrograph aboard the Hubble Space Tele-
scope [21.24].
21.7.2 Hyperfine Effects
The magnetic hyperfine structure (hfs) is due to an in-
teraction between the magnetic field generated by the
electrons and the nuclear magnetic dipole moment. The
interaction couples the nuclear and electronic angular
momenta to a total momentum F = I+ J, and the in-
teraction energy can be written as the expectation value
of a scalar product between an electronic and a nuclear
tensor operator [21.25] (see Chapt. 16)
W
M1
(J ) =
γ
I
γ
J
IJFM
F
| T
(1)
·M
(1)
| γ
I
γ
J
IJFM
F
.
(21.62)
The nuclear operator M
(1)
is related to the scalar mag-
netic dipole moment, µ
I
, according to
γ
I
II | M
(1)
0
| γ
I
II
= µ
I
. (21.63)
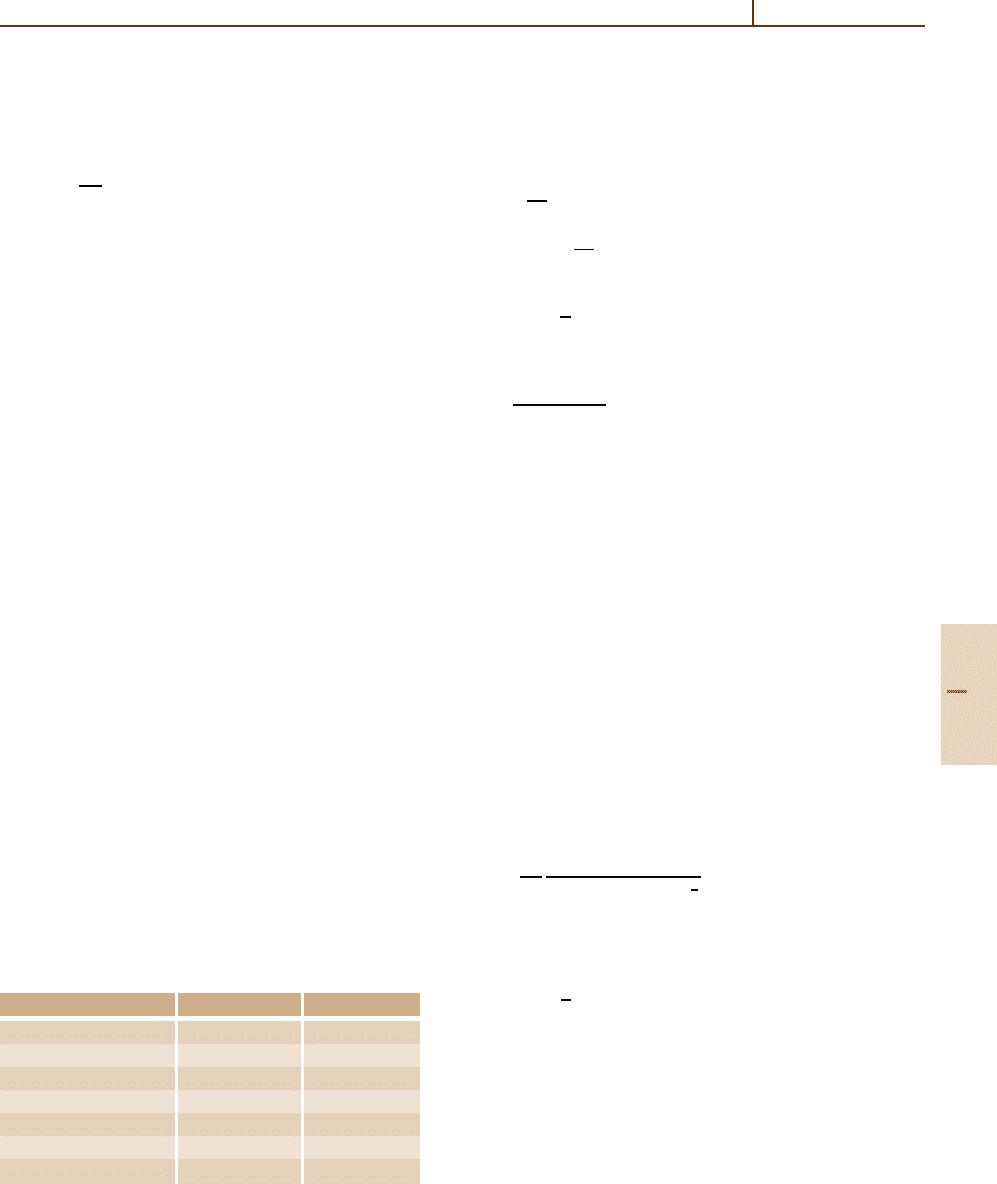
Table 21.4 Specific mass shift parameter and electron den-
sity at the nucleus as a function of the active set
Active set S(a.u.) |ψ(0)|
2
HF 0.000 00 72.629
2s 1p −0.020 17 72.452
3s 2p 1d 0.625 18 72.490
4s 3p 2d 1f 0.624 81 72.497
5s 4p 3d 2f 1g 0.601 69 72.501
6s 5p 4d 3f 2g 1h 0.598 03 72.503
7s 6p 5d 4f 3g 2h 1i 0.597 09 72.504
The magnetic dipole moments are known quantities, ob-
tained with high accuracy from experiments. For a recent
tabulation see [21.26].
The electronic operator is
T
(1)
=
α
2
2
N
i=1
2l
(1)
(i)r
−3
i
−g
s
√
10
C
(2)
(i) ×s
(1)
(i)
(1)
r
−3
i
+g
s
8
3
πδ(r
i
)s
(1)
(i)
, (21.64)
where g
s
= 2.002 3193 is the electron spin g-fac-
tor, δ(r) the three-dimensional delta function and
C
(k)
q
=
√
4π/(2k +1)Y
kq
, with Y
kq
being a normalized
spherical harmonic. The first term of the electronic op-
erator represents the magnetic field generated by the
orbiting electric charges and is called the orbital term.
The second term represents the field generated by the
orbiting magnetic dipole moments, which are coupled
to the spin of the electrons. This is the spin-dipole term.
The last term represents the contact interaction between
the nuclear magnetic dipole moment and the electron
magnetic moment. It is called the Fermi contact term
and contributes only for s-electrons.
By recoupling I and J, the interaction energy can be
rewritten as
W
M1
(J ) =(−1)
I+J−F
W(IJIJ; F1)
×γ
J
JT
(1)
γ
J
Jγ
I
IM
(1)
γ
I
I ,
(21.65)
where W(IJIJ; F1) is a W coefficient of Racah. When
the magnetic dipole interaction constant
A
J
=
µ
I
I
1
[J(J +1)(2J +1)]
1
2
γ
J
JT
(1)
γ
J
J
(21.66)
is introduced, the energy is given by
W
M1
(J ) =
1
2
A
J
C , (21.67)
where C = F(F +1) − J(J +1) − I(I +1). In theoret-
ical studies, the A-factor is often given as a linear
combination of the hyperfine parameters
a
l
=γLSM
L
M
S
|
i
l
(1)
0
(i)r
−3
i
| γLSM
L
M
S
,
(21.68)
Part B 21.7
320 Part B Atoms
a
d
=γLSM
L
M
S
|
i
2C
(2)
0
(i)s
(1)
0
(i)r
−3
i
| γLSM
L
M
S
, (21.69)
a
c
=γLSM
L
M
S
|
i
2s
(1)
0
(i)r
−2
i
δ(r
i
) | γLSM
L
M
S
, (21.70)
where M
L
= L and M
S
= S. These parameters corre-
spond to the orbital, spin-dipole and Fermi contact term
of the electronic operator.
The electric hyperfine structure is due to the interac-
tion between the electric field gradient produced by the
electrons and the nonspherical charge distribution of the
nucleus. The interaction energy is
W
E2
(J ) =γ
I
γ
J
IJFM
F
| T
(2)
·M
(2)
| γ
I
γ
J
IJFM
F
, (21.71)
where the nuclear operator M
(2)
is related to the scalar
electric quadrupole moment, Q, according to
γ
I
II | M
(2)
0
| γ
I
II=
Q
2
.
(21.72)
The electronic operator is
T
(2)
=−
N
i=1
C
(2)
(i)r
−3
i
, (21.73)
and represents the electric field gradient. By introducing
the electric quadrupole interaction constant B,
B
J
=2Q
J(2J −1)
(J +1)(2J +1)(2J +3)
1
2
×γ
J
JT
(2)
γ
J
J , (21.74)
the interaction energy can be written as
W
E2
(J ) = B
J
3
4
C(C +1) − I(I +1)J(J +1)
2I(2I −1)J(2J −1)
.
(21.75)
In many cases the electric hyperfine interaction is
weaker than the magnetic and manifests itself as a small
deviation from the Landé interval rule for the magnetic
hfs. If the electronic part of the interaction can be cal-
culated accurately, a value of the electric quadrupole
moment Q, which is a difficult quantity to measure
with direct nuclear techniques, can be deduced from
the measured B-factor. A recent tabulation of nuclear
quadrupole moments is given in [21.27].
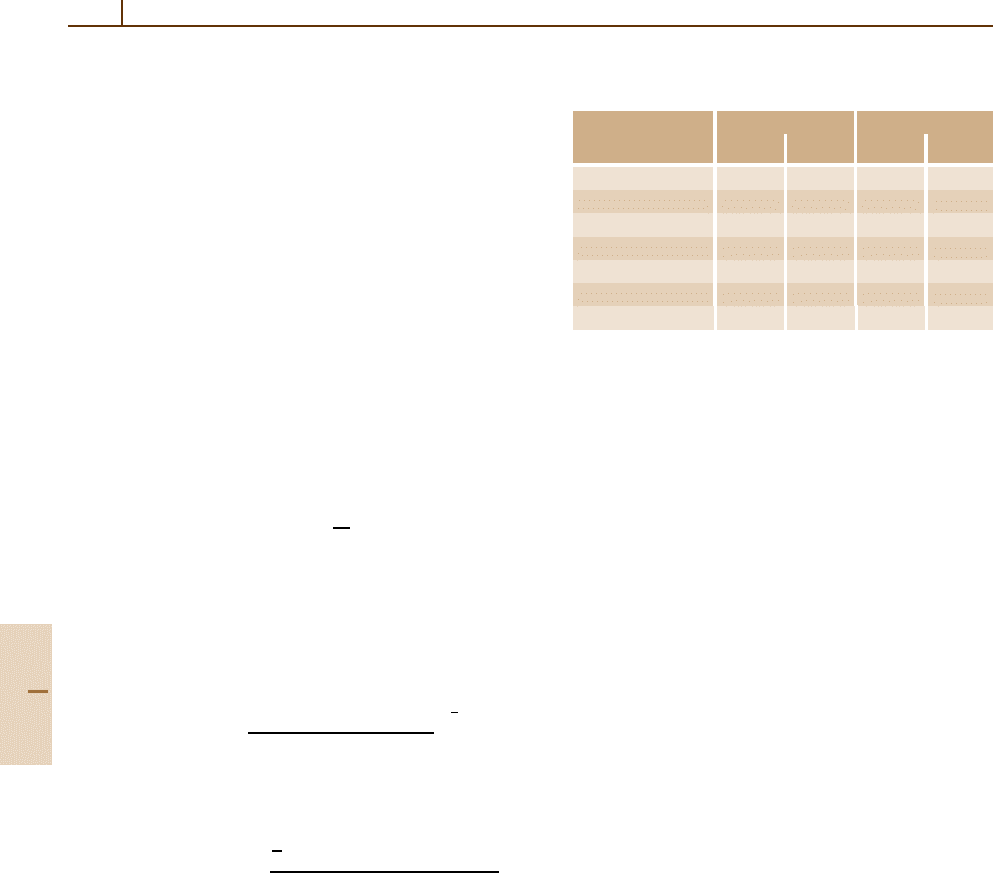
Table 21.5 shows the convergence of an MCHF ac-
tive space calculation for two different isotopes for the
1s
2
2s2p
1
P state of B II [21.24]. Some oscillations are
Table 21.5 MCHF Hyperfine constants (in MHz) for the
1s
2
2s2p
1
P state of B II
10
B
11
B
Active set
A
1
B
1
A
1
B
1
HF 60.06 8.338 179.36 4.001
2s 1p 60.22 8.360 179.83 4.011
3s 2p 1d 60.98 8.193 182.11 3.932
4s 3p 2d 1f 60.05 7.677 179.34 3.684
5s 4p 3d 2f 1g 60.48 7.764 180.62 3.725
6s 5p 4d 3f 2g 1h 60.85 8.052 181.71 3.864
7s 6p 5d 4f 3g 2h 1i 60.81 8.002 181.60 3.840
observed since each new “layer” of orbitals may localize
in different regions of space.
21.7.3 Metastable States and Lifetimes
States above an ionization threshold may decay via
a radiationless transition to a continuum. When the inter-
action with the continuum is spin-forbidden the state is
metastable. The nsnp
24
P of negative ions, for example,
decay through Breit–Pauli interactions with nsnp
2
dou-
blets. The latter, in turn interact with continuum states,
thus opening a decay channel. Such metastable states
may be treated as bound states.
The foundation for the theory of autoionization was
laid down by Fano [21.28], where he developed a con-
figuration interaction (CI) theory for autoionization. Let
Ψ
b
(N;γLS) be a normalized, multiconfiguration com-
ponent of a discrete perturbor for an N-electron system,
in which all orbitals are bound orbitals, decaying ex-
ponentially for large r.LetΨ
k
be an asymptotically
normalized continuum component of the wave func-
tion, also for an N-electron system, at energy E,of
the form,
Ψ
k
(N; Eγ
LS) =|Ψ
b
N −1;β
˜
L
˜
S
· φ(kl)LS ,
(21.76)
where Ψ
b
N −1;β
˜
L
˜
S
is a bound MCHF wave function
for the (N −1)-electron target system. Then, in the LS-
coupling scheme, the width of the autoionizing state is
given by the “Golden Rule”
Γ = 2πV
2
E
0
, (21.77)
where
V
E
0
=Ψ
b
(N;γLS)|H −E
0
|Ψ
k
(N; E
0
γ
LS) .
(21.78)
A similar formula can be derived for the LSJ-scheme
and the Breit–Pauli Hamiltonian. In the above equation,
Part B 21.7

Atomic Structure: Multiconfiguration Hartree–Fock Theories 21.7 Atomic Properties 321
E
0
=Ψ
b
(N;γLS)|H|Ψ
b
(N;γLS). The energy of the
core, or target, is E
target
=
Ψ
b
N −1;β
˜
L
˜
S
|H|Ψ
b
N −
1;β
˜
L
˜
S
,whereH in the latter equation is the Hamilto-
nian for an (N −1)-electron system.
The wave function for the continuum compo-
nent is assumed to have only one unknown, namely
φ(kl) = P
kl
(r)|ls, the one-particle continuum function,
where |ls is the known spin–angular part. The radial
equation for P
kl
(r) has exactly the same form as (21.26),
except that ε
nl,nl
=−k
2
,whereE
0
= E
target
+k
2
/2.
The radial function can be obtained iteratively using an
SCF procedure from outward integration.
One of the more accurate calculations of a lifetime
is that for He
−
1s2s2p
4
P
5/2
by Miecznik et al. [21.29].
In this case, a lifetime of (345±10) µs was found
and compared with a recent experimental value of
(350±15) µs [21.30]. In this LSJ state, the
4
Pinter-
acts with the 1s
2
k f
2
F. It was found that correlation in
the target of the continuum orbital modified the life-
time. In calculations like these, it is always a question
whether orthogonality conditions should be applied [as
in projection operator formalism (see Chapt. 25)]. Some
theorems relating to this question have been published
by Brage et al. [21.31].
The position and widths of autoionizing resonances
can also be determined from the study of photoioniza-
tion or photodetachment using a spline Galerkin method
together with inverse iteration. No boundary condition
need be applied nor is there an inner and outer re-
gion. Resonance properties are obtained from a fitting of
the cross-section [21.32]. Non-orthogonal, spline-based
R-matrix methods with an inner and outer region have
also been developed where there is no need of the “Buttle
correction” and, at the same time, the non-orthogonality
eliminates the need of certain pseudo-states [21.33].
For an extensive review of the application of splines
in atomic and molecular physics, see [21.34].
21.7.4 Transition Probabilities
The most fundamental quantity for the probability of
a transition from an initial state i to a final state f is the
reduced matrix element related to the line strength by
S
1/2
=Ψ
i
||O||Ψ
f
, (21.79)
where O is the transition operator. In the case of the
electric dipole transition, there are two frequently used
forms: the length form O =
j
r
j
, and the velocity
form, O =
j
∇
j
/E
if
,whereE
if
= E
f
−E
i
.Forex-
act non-relativistic wave functions, the two forms are
equivalent, but for approximate wave functions, the ma-
trix elements in general differ. Thus, the computation of
the line strength and the oscillator strength, or f -value,
where
f = (2/3)E
if
S/[(2S
i
+1)(2L
i
+1)] ,
forms a critical test of the wave function in non-
relativistic theory and also the model describing
a many-electron system. The same operators are often
also used in Breit–Pauli calculations. In this case, the
velocity form of the operator has neglected some terms
of order (αZ)
2
and the length value is preferred.
Some well-known discrepancies between theory and
experiment existed for more than a decade for the res-
onance transitions of Li and Na. For the nonrelativistic
2s–2p transition in Li, a full-core CI [21.35] calcula-
tion produced f -values of (0.747 04, 0.747 04, 0.753 78)
for the length-, velocity-, and acceleration form, respec-
tively. When relativistic corrections were included, the
value changed to 0.747 15, in agreement with a num-
ber of theories, tabulated to only four decimal places.
A fast beam-laser experiment by Gaupp et al. [21.36],
yielded a value of 0.7416±0.0012 but this value was
revised in 1996 by a beam-gas-laser experiment in per-
fect agreement with theory [21.37]. In the case of the
resonance transition in Na, when theory included cor-
relation in the core as well as core-polarization and
some relativistic effects, results were in agreement with
an almost simultaneous cascade of new experimental
values [21.38].
For MCHF calculations of transition data, an impor-
tant consideration is that the matrix element is between
two different states. For independently optimized wave
functions, the orbitals of the initial and final states are
not orthonormal, as assumed when Racah algebra tech-
niques are used to evaluate the transition matrix element.
Through the use of biorthogonal transformations, the
orbitals and the coefficients of expansion of the wave
functions of the initial and final state can be transformed
efficiently so that standard Racah algebra techniques
may be applied [21.39]. Table 21.6 shows the conver-
gence of an MCHF calculation for the ground state of
Boron from independently optimized wave functions.
21.7.5 Electron Affinities
By definition, the electron affinity is A
e
= E(A
−
) −
E(A). Thus it is the energy difference between Hamilto-
nians differing by one electron. Correlation plays a very
important role in the binding of the extra electron in
the negative ion. It has been known for a long time that
the alkali metals have a positive electron affinity, but
Part B 21.7
322 Part B Atoms
only recently has it been found, theoretically [21.40]
and experimentally [21.41], that some of the alkaline
earths may also be able to bind an extra electron. The
d-electrons need to play a strong role, so Be and Mg,
do not have a positive A
e
, but according to the most
recent experimental measurement, the electron affin-
ity for Ca is 18 meV [21.42]. A calculation based on
the spline methods and using a model potential with
adjustable parameters to describe the core, obtained
a value of 17.7 meV [21.43] in close agreement with
experiment.
Atomic systems such as Ca are often thought of as
two-electron systems and indeed, for qualitative descrip-
tions, many observations can be explained. A number of
physical effects need to be considered when predicting
such electron affinities:
(i) Valence correlation is crucial for obtaining binding.
(ii) Intershell correlation between the valence electrons
and the core (core polarization) is also important. The
first electron may polarize the core considerably, but
this is reduced by the second electron since the two
avoid each other dynamically and prefer to be on oppo-
site sides of the core.
(iii) Core rearrangement, which occurs because of the
presence of one or more outer electrons, and is particu-
larly large if any of these penetrate the core. In the case of
Ca
+
, the fixed-core Hartree–Fock energy of 3d
2
D state
is 300 meV higher if computed in the fixed potential the
Ca
+2
ion compared with a fully variational calculation!
(iv) Intracore exclusion effects due to the presence of an
Table 21.6 Convergence of transition data for the
1s
2
2s
2
2p
2
P
o
→ 1s
2
2s2p
22
D transition in Boron with in-
creasing active set
N gf
l
gf
v
S
l
∆E (cm
−1
)
3 0.6876 0.8156 2.5534 53 197
4 0.2456 0.2696 0.9959 48 720
5 0.2625 0.2695 1.0705 48 440
6 0.2891 0.2866 1.1868 48 125
7 0.2928 0.2900 1.2036 48 051
Expt.
a
0.28(02) 47 857
a
[21.44]
extra valence electron which reduces the correlation of
the core.
(v) Relativistic shift effects, which are present in
observed levels and are particularly important for
s-electrons.
Model potential methods attempt to capture all but
valence correlation in a potential so that calculations
for Calcium, for example, can proceed as though for
a two-electron system. A review of various theoretical
approaches, many of which include different effects,
may be found in [21.45].
For small systems such as Li, the electron affinity has
been computed [21.46] to experimental accuracy [21.47]
of (0.6176 ±0.0002) eV. In neutral oxygen, it has been
found that there is an isotope effect on the electron
affinity [21.48].
21.8 Summary
More comprehensive treatments of atomic structure
may be found [21.49, 50]. An atomic structure pack-
age is available for many of the calculations described
here [21.51]. A review of the application of systematic
procedures to the prediction of atomic properties has
been published [21.52,53].
References
21.1 R. Glass, A. Hibbert: Comput. Phys. Commun. 16,19
(1978)
21.2 I. Lindgren, J. Morrison: Atomic Many-Body Theory
(Springer, Berlin, Heidelberg, New York 1982)
21.3 P.-O. Löwdin: Phys. Rev. 97, 1509 (1955)
21.4 D. Layzer, Z. Horák, M. N. Lewis, D. P. Thompson:
Ann. Phys. (N. Y.) 29,101(1964)
21.5 D. Layzer: Ann. Phys. (N. Y.) 8, 271 (1959)
21.6 C. A. Nicolaides, D. R. Beck: Chem. Phys. Lett. 35,79
(1975)
21.7 C. Froese Fischer: Comput. Phys. Rep. 3,273(1986)
21.8 C. Froese Fischer: J. Comput. Phys. 13, 502 (1973)
21.9 M. R. Godefroid, J. Lievin, J.-Y. Metz: Int. J. Quan-
tum Chem. XL, 243 (1991)
21.10 G. Tachiev, C. Froese Fischer: J. Phys. B 32,5805
(1999)
21.11 S. A. Alexander, J. Olsen, P. Öster, H. M. Quiney,
S. Salomonson, D. Sundholm: Phys. Rev. A 43, 3355
(1991)
21.12 C. Froese Fischer: J. Phys. B 26, 855 (1993)
21.13 K. T. Chung, X.-W. Zhu, Z.-W. Wang: Phys. Rev. A
47, 1740 (1993)
Part B 21

Atomic Structure: Multiconfiguration Hartree–Fock Theories References 323
21.14 E. Lindroth, H. Persson, S. Salomonson,
A.-M. Mårtensson-Pendrill: Phys. Rev. A 45,1493
(1992)
21.15 R. L. Kelly: J. Phys. Chem. Ref. Data 16, 1371–1678
(1987), Suppl. 1
21.16 K. T. Chung: Many-Body Theory of Atomic Struc-
ture and Photoionization, ed. by T. N. Chang (World
Scientific, Singapore 1993) p. 83
21.17 E. Clementi, C. Roetti: At. Data Nucl. Data Tables 14,
177 (1974)
21.18 A. Hibbert: Comput. Phys. Commun. 9,141(1975)
21.19 M. Bentley, H. W. van der Hart, M. Landtman,
G. M. S. Lister, Y.-T. Shen, N. Vaeck: Phys. Scr. T47,
7 (1993)
21.20 T. Brage, C. Froese Fischer: Phys. Scr. 49, 651 (1994)
21.21 O. Zatsarinny, C. Froese Fischer: J. Phys. B 35, 4669
(2002)
21.22 D. S. Hughes, C. Eckart: Phys. Rev. 36, 694 (1930)
21.23 W. H. King: Isotope Shifts in Atomic Spectra
(Plenum, New York 1984)
21.24 P. Jönsson, S. G. Johansson, C. Froese Fischer:
Astrophys. J. 429, L45 (1994)
21.25 I. Lindgren, A. Rosén: Case Stud. At. Phys. 1772 4,
93 (1974)
21.26 P. Raghavan: At. Data Nucl. Data Tables 42,189
(1989)
21.27 P. Pyykkö: Z. Naturforsch. 47a, 189 (1992)
21.28 U. Fano: Phys. Rev. 129, 1866 (1961)
21.29 G. Miecznik, T. Brage, C. Froese Fischer: Phys. Rev.
A 47, 3718 (1993)
21.30 T. Andersen, L. H. Andersen, P. Balling, H. K. Hau-
gen, P. Hvelplund, W. W. Smith, K. Taulbjerg: Phys.
Rev. A 47, 890 (1993)
21.31 T. Brage, C. Froese Fischer, N. Vaeck: J. Phys. B 26,
621 (1993)
21.32 J. Xi, C. Froese Fischer: Phys. Rev. A 53, 3169
(1996)
21.33 O. Zatsarinny, C. Froese Fischer: J. Phys. B 35, 4161
(2002)
21.34 H. Bachau, E. Cormier, J. E. Hansen, F. Martin: Rep.
Prog. Phys. 64, 1815 (2001)
21.35 K. T. Chung: Proceedings of the international con-
ference on highly charged ions, Manhattan Kansas,
1992. In: AIP Conference Proceedings #274 (AIP, New
York 1993)
21.36 A. Gaupp, P. Kuske, H. J. Andrä: Phys. Rev. A 26,
3351 (1982)
21.37 U. Volz, H. Schmoranzer: Phys. Scripta T65,48
(1996)
21.38 P. Jönsson, A. Ynnerman, C. Froese Fischer,
M. R. Godefroid, J. Olsesn: Phys. Rev. A 53,4021
(1996)
21.39 J. Olsen, M. R. Godefroid, P. Jönsson, P. Å. Malm-
qvist, C. Froese Fischer: Phys. Rev. E 52, 449 (1995)
21.40 C. Froese Fischer, J. B. Lagowski, S. H. Vosko: Phys.
Rev. Lett. 59, 2263 (1987)
21.41 D. J. Pegg, J. S. Thompson, R. N. Compton, G. D. Al-
ton: Phys. Rev. Lett. 59, 2267 (1989)
21.42 K. W. McLaughlin, D. W. Duquette: Phys. Rev. Lett.
72, 1176 (1994)
21.43 H. W. van der Hart, C. Laughlin, J. E. Hansen: Phys.
Rev. Lett. 71, 1506 (1993)
21.44 T. R. O’Brian, J. E. Lawler: Astron. Astrophys. 255,
420 (1992)
21.45 C. Froese Fischer, T. Brage: Can. J. Phys. 70, 1283
(1992)
21.46 C. Froese Fischer: J. Phys. B 26, 855 (1993)
21.47 J. Dellwo, Y. Liu, D. J. Pegg, G. D. Alton: Phys. Rev.
A 45, 1544 (1992)
21.48 M. R. Godefroid, C. Froese Fischer: Phys. Rev. A 60,
R2637 (1999)
21.49 I. I. Sobel’man: Introduction to the Theory of
Atomic Spectra (Pergamon, Oxford 1972)
21.50 R. D. Cowan: The Theory of Atomic Structure and
Spectra (Univ. of California Press, Berkeley 1981)
21.51 C. Froese Fischer: Computer Phys. Commun. 64,369
(1991)
21.52 C. Froese Fischer, P. Jönsson: Comput. Phys. Com-
mun. 84, 37 (1994)
21.53 C. Froese Fischer, T. Brage, P. Jönsson: Computa-
tional Atomic Structure: An MCHF approach (IOP,
Bristol 1997)
Part B 21
325
Relativistic Ato
22. Relativistic Atomic Structure
Relativistic quantum mechanics is required for
the description of atoms and molecules whenever
their orbital electrons probe regions of space with
high potential energy near the atomic nuclei.
Primary effects of a relativistic description include
changes to spatial and momentum distributions;
spin–orbit interactions; quantum electrodynamic
corrections such as the Lamb shift; and vacuum
polarization. Secondary effects in many-electron
systems arise from shielding of the outer electrons
by the distributions of electrons in penetrating
orbitals; they change orbital binding energies and
dimensions and so modify the order in which
atomic shells are filled in the lower rows of the
Periodic Table.
Relativistic atomic and molecular structure
theory can be regarded as a simplification of the
fundamental description provided by quantum
electrodynamics (QED). This treats the atom
or molecule as an assembly of electrons and
atomic nuclei interacting primarily by exchanging
photons. This model is far too difficult and
general for most purposes, and simplifications
are required. The most important of these is the
representation of the nuclei as classical charge
distributions, or even as point particles. Since the
motion of the nuclei is usually slow relative to the
electrons, it is often adequate to treat the nuclear
motion nonrelativistically, or even to start from
nuclei in fixed positions, correcting subsequently
for nuclear motion.
The emphasis in this chapter is on relativistic
methods for the calculation of atomic structure
for general many-electron atoms based on
an effective Hamiltonian derived from QED in
the manner sketched in Sect. 22.2 below. An
understanding of the Dirac equation, its solutions
and their numerical approximation, is essential
material for studying many-electron systems,
just as the corresponding properties of the
Schrödinger equation underpin Chapt. 21.We
shall use atomic units throughout. Where it aids
interpretation we shall, however, insert factors
of c, m
e
and . In these units, the velocity of
light, c, has the numerical value
α
−1
= 137.035 999 11(46),whereα is the fine
structure constant.
22.1 Mathematical Preliminaries.................. 326
22.1.1 Relativistic Notation:
Minkowski Space-Time .............. 326
22.1.2 Lorentz Transformations............. 326
22.1.3 Classification of Lorentz
Transformations........................ 326
22.1.4 Contravariant and Covariant
Vectors..................................... 327
22.1.5 Poincaré Transformations........... 327
22.2 Dirac’s Equation .................................. 328
22.2.1 Characterization of Dirac States... 328
22.2.2 The Charge-Current 4-Vector ...... 328
22.3 QED: Relativistic Atomic
and Molecular Structure ....................... 329
22.3.1 The QED Equations of Motion ...... 329
22.3.2 The Quantized Electron–Positron
Field........................................ 329
22.3.3 Quantized Electromagnetic Field . 330
22.3.4 QED Perturbation Theory ............ 331
22.3.5 Propagators.............................. 333
22.3.6 Effective Interaction of Electrons . 333
22.4 Many-Body Theory For Atoms ............... 334
22.4.1 Effective Hamiltonians ............... 335
22.4.2 Nonrelativistic Limit:
Breit–Pauli Hamiltonian ............ 335
22.4.3 Perturbation Theory:
Nondegenerate Case.................. 335
22.4.4 Perturbation Theory:
Open-Shell Case........................ 336
22.4.5 Perturbation Theory: Algorithms . 337
22.5 Spherical Symmetry ............................. 337
22.5.1 Eigenstates of Angular
Momentum .............................. 337
22.5.2 Eigenstates of Dirac Hamiltonian
in Spherical Coordinates ............ 338
22.5.3 Radial Amplitudes ..................... 340
22.5.4 Square Integrable Solutions........ 341
22.5.5 Hydrogenic Solutions ................. 342
22.5.6 The Free Electron Problem
in Spherical Coordinates ............ 343
Part B 22

326 Part B Atoms
22.6 Numerical Approximation
of Central Field Dirac Equations ............ 344
22.6.1 Finite Differences ...................... 344
22.6.2 Expansion Methods ................... 345
22.6.3 Catalogue of Basis Sets
for Atomic Calculations .............. 347
22.7 Many-Body Calculations ....................... 350
22.7.1 Atomic States............................ 350
22.7.2 Slater Determinants................... 350
22.7.3 Configurational States................ 350
22.7.4 CSF Expansion ........................... 350
22.7.5 Matrix Element Construction....... 350
22.7.6 Dirac–Hartree–Fock
and Other Theories .................... 351
22.7.7 Radiative Corrections ................. 353
22.7.8 Radiative Processes ................... 353
22.8 Recent Developments........................... 354
22.8.1 Technical Advances ................... 354
22.8.2 Software for Relativistic Atomic
Structure and Properties ............ 354
References .................................................. 355
22.1 Mathematical Preliminaries
22.1.1 Relativistic Notation:
Minkowski Space-Time
An event in Minkowski space-time is defined by
a 4-vector x ={x
µ
} (µ = 0, 1, 2, 3) where x
0
= ct is
the time coordinate and x
1
, x
2
, x
3
are Cartesian coordi-
nates in 3-space. The bilinear form (The Einstein suffix
convention, in which repeated pairs of Greek subscripts
are assumed to be summed over all values 0, 1, 2, 3, will
be used where necessary in this chapter.)
(x, y) = x
µ
g
µν
y
ν
, (22.1)
in which
g =
g
µν
=
g
µν
=
1000
0 −100
00−10
000−1
(22.2)
are called metric coefficients, defines the metric of
Minkowski space.
22.1.2 Lorentz Transformations
Lorentz transformations are defined as linear map-
pings Λ such that
(Λx,Λy) = (x, y)
(22.3)
so that
g
µν
= Λ
ρ
µ
g
ρσ
Λ
σ
ν
. (22.4)
This furnishes 10 equations connecting the 16 compo-
nents of Λ; at most 6 components can be regarded as
independent parameters. The (infinite) set of Λ matri-
ces forms a regular matrix group (with respect to matrix
multiplication) called the Lorentz group, L, designated
O(3,1) [22.1, 2].
22.1.3 Classification of Lorentz
Transformations
Rotations
Lorentz transformations with matrices of the form
Λ =
1 0
0 R
,
(22.5)
where R ∈ SO(3) is an orthogonal 3 × 3 matrix with de-
terminant +1, and 0 is a null three dimensional column
vector, correspond to three-dimensional space rotations.
They form a group isomorphic to SO(3).
Boosts
Lorentz transformations with matrices of the form
Λ =
γ(v) γ(v)v
γ(v)v I
3
+(γ(v) −1)nn
,
(22.6)
with v = vn a three dimensional column vector, |n|=1,
v =|v| and γ(v) = (1 −v
2
/c
2
)
−1/2
, are called boosts.
The matrix Λ describes an ‘active’ transformation from
an inertial frame in which a free classical particle is at
rest to another inertial frame in which its velocity is v.
Boosts form a submanifold of L though they do not
in general form a subgroup. However, the set of boosts
in a fixed direction n form a one-parameter subgroup.
Discrete Transformations
The matrices
P =
1 0
0 −I
3
, T =
−1 0
0 I
3
with PT =−I
4
(22.7)
are called discrete Lorentz transformations and form
a subgroup of the Lorentz group along with the iden-
Part B 22.1